The search for new materials with enhanced mechanical properties has previously been constrained by the sheer complexity of the compositional design space. Multi-principal element alloys offer exceptional strength at high temperatures but present formidable challenges for traditional design approaches.
Researchers Alireza Ghafarollahi and Markus Buehler at Massachusetts Institute of Technology continue their work into autonomous AI discovery, with the introduction of a transformative framework that combines graph neural networks with large language model-powered multi-agent systems to accelerate the discovery and optimization of these advanced materials.
The work addresses a fundamental bottleneck in materials science. While theoretical models can predict macroscopic properties like yield stress from atomic-scale parameters such as the Peierls barrier and solute-screw interaction energy, computing these parameters traditionally requires expensive atomistic simulations that can take days or months when exploring vast compositional spaces.
The authors demonstrate how a carefully designed graph neural network can predict these critical quantities in seconds, and how integrating this capability into a multi-agent artificial intelligence system creates an automated platform for rapid alloy design and analysis.
This work builds on the discoveries made by Buehler's team in the realm of Co-Scientist AI. Read more at PRefLexOR, Plant Materials, SPARKS, and GraphPreFLexOR.
Key Takeaways
- A graph neural network trained on body-centered cubic alloy structures predicts the Peierls barrier with a mean absolute error of just 37 millielectronvolts and potential energy changes with 60 millielectronvolts error, enabling rapid exploration of compositional space without costly nudged elastic band calculations.
- The multi-agent system integrates advanced reasoning models from the GPT family, specialized AI agents with distinct roles, and the graph neural network to automate complex multifaceted alloy design problems including yield stress prediction across temperature ranges.
- Despite training on a relatively small subset of the ternary niobium-molybdenum-tantalum compositional space, the model accurately generalizes to unseen compositions, demonstrating the power of graph-based representations for capturing atomic-scale defect physics.
- The framework reduces the computational time for exploring 231 compositions in the ternary space from months to seconds, revealing nonlinear trends in both the Peierls barrier and solute-dislocation interaction parameters across binary and ternary systems.
- Integration of physics-based solute-strengthening theories with machine learning predictions enables the automated calculation of temperature-dependent yield stress, validated against experimental data for various alloy compositions.
- The system demonstrates human-in-the-loop functionality, allowing iterative refinement and follow-up queries that enable deeper exploration of specific compositional regions or material behaviors.
Bridging Scales with Intelligent Automation
Materials discovery has entered a new era where the integration of deep learning, physics-based theories, and intelligent automation enables unprecedented exploration of design spaces. The latest framework presented by Ghafarollahi and Buehler exemplifies this transformation.
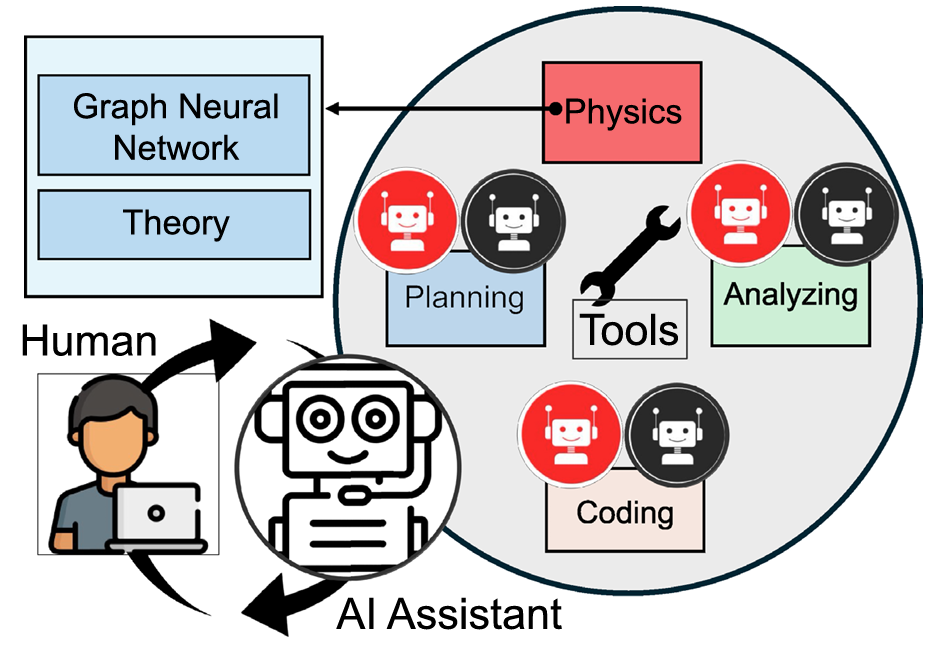
Figure 4. Overview of the graph neural network (GNN)-powered, large language model based multi-agent system developed here. The system consists of a human and an artificial intelligence (AI) assistant agent at its core, where the human poses queries, and the AI assistant provides responses, seamlessly steering the problem-solving process with the help of integrated tools. These tools are responsible for various tasks, including planning, coding, and multimodal analysis, and each incorporates a set of AI agents that dynamically collaborate to solve complex tasks. A key component is the phys ics tool, which includes newly developed GNN models to retrieve essential physical parameters (such as the Peierls barrier and potential energy change) as well as physics based theories (such as solute-strengthening theory). The GNN models enable the rapid prediction of fundamental materials properties, bypassing the need for costly atomistic simulations. The iterative collaboration between agents within the tools and the seam less interaction between the human and AI assistant allows for efficient resolution of complex materials design challenges. Credit: Buehler et al
At its core lies a graph neural network architecture that learns to map atomic configurations containing screw dislocations directly to fundamental materials properties. Unlike traditional surrogate models that may sacrifice accuracy for speed, this approach achieves both by leveraging the Principal Neighborhood Aggregation graph convolution operator (Corso et al., 2020), which has demonstrated superior performance on graph regression tasks through its combination of multiple aggregators including mean, maximum, minimum, and standard deviation operations with degree-based scalers.
The graph representation itself is elegant in its construction. Each atomic configuration is represented as a graph where nodes correspond to atoms within a cylindrical region of 16 angstroms radius centered at the screw dislocation core, and edges connect atoms within a 2.8 angstrom cutoff distance.
Graph node features encode both chemical information through one-hot encoding of solute types and structural information through the z-component of the screw dislocation displacement field calculated for pure molybdenum.
Critically, the structural feature remains constant across all compositions, eliminating the need for atomic relaxation during inference and enabling the model to predict properties for new random configurations instantaneously.
This graph neural network forms one component of a larger multi-agent ecosystem. The system builds upon AtomAgents, a multimodal multi-agent framework previously developed for extracting physical insights from atomistic simulations (Ghafarollahi and Buehler, 2025).
In this enhanced version, the graph neural network replaces computationally prohibitive simulations for critical parameter predictions, while the multi-agent architecture orchestrates the entire design process.
The User agent poses queries, the AI Assistant coordinates responses using various tools, and specialized autonomous agents collaborate to tackle different aspects of the problem.
A planning tool driven by planner and reviewer agents generates detailed step-by-step strategies, while a coding tool writes Python scripts for visualization, and physics tools leverage both the graph neural network and established theoretical frameworks like the Maresca-Curtin solute-strengthening theory.
The Maresca-Curtin theory deserves particular attention as it connects atomic-scale features to macroscopic yield stress in body-centered cubic alloys (Maresca and Curtin, 2020). The theory considers three strengthening mechanisms related to screw dislocation motion: the Peierls-like mechanism representing intrinsic lattice resistance, the kink glide mechanism describing how kinks propagate along the dislocation line, and cross-kink formation and unpinning.
Each mechanism contributes to the overall temperature and strain-rate dependent yield stress. The graph neural network's ability to rapidly compute the Peierls barrier and solute-screw interaction energy parameter provides two of the most computationally expensive inputs to this theory, enabling automated yield stress predictions that would otherwise require prohibitive computational resources.
Transformative Impact on Materials Design
The significance of this work extends well beyond the specific alloy system studied. Multi-principal element alloys represent a massive unexplored compositional space with potential for discovering materials with unprecedented combinations of properties.
Traditional approaches to exploring this space have faced a fundamental tradeoff between accuracy and computational efficiency. High-fidelity methods like density functional theory or molecular dynamics with machine-learned potentials can capture the physics accurately but become impractical when screening thousands of compositions. Simplified models or linear mixing rules can enable broader screening but often miss critical nonlinear effects that govern material behavior.
The graph neural network approach demonstrated here offers a compelling middle ground. By training on a carefully curated dataset the model learns to capture the complex relationship between random solute environments and dislocation energetics.
The statistical nature of the problem makes this particularly challenging since random fluctuations in solute distribution around the dislocation core can significantly affect material properties. Yet the model achieves strong performance even for compositions far from the training data, including binary systems and the equimolar niobium-molybdenum-tantalum composition.
From a broader materials science perspective, this framework addresses a critical gap in inverse design. While many machine learning approaches excel at forward prediction, they struggle to incorporate the multiscale, multimodal knowledge required for true materials discovery.
The multi-agent architecture enables precisely this integration. It can reason across different scales, from atomistic features to macroscopic properties. It can incorporate diverse data types, from simulation results to experimental measurements to theoretical models. Most importantly, it can adapt to new information and user feedback, making it a true collaborator rather than just a regression tool.
The cross-domain applicability of this approach also merits emphasis. While demonstrated for body-centered cubic refractory alloys, the methodology extends naturally to other crystalline structures including face-centered cubic and hexagonal close-packed systems, and to other types of defects beyond screw dislocations.
The graph representation framework is material-agnostic, and the multi-agent architecture can be reconfigured for different design objectives or materials classes, positioning the system as a general platform for automated materials discovery across multiple disciplines.
.
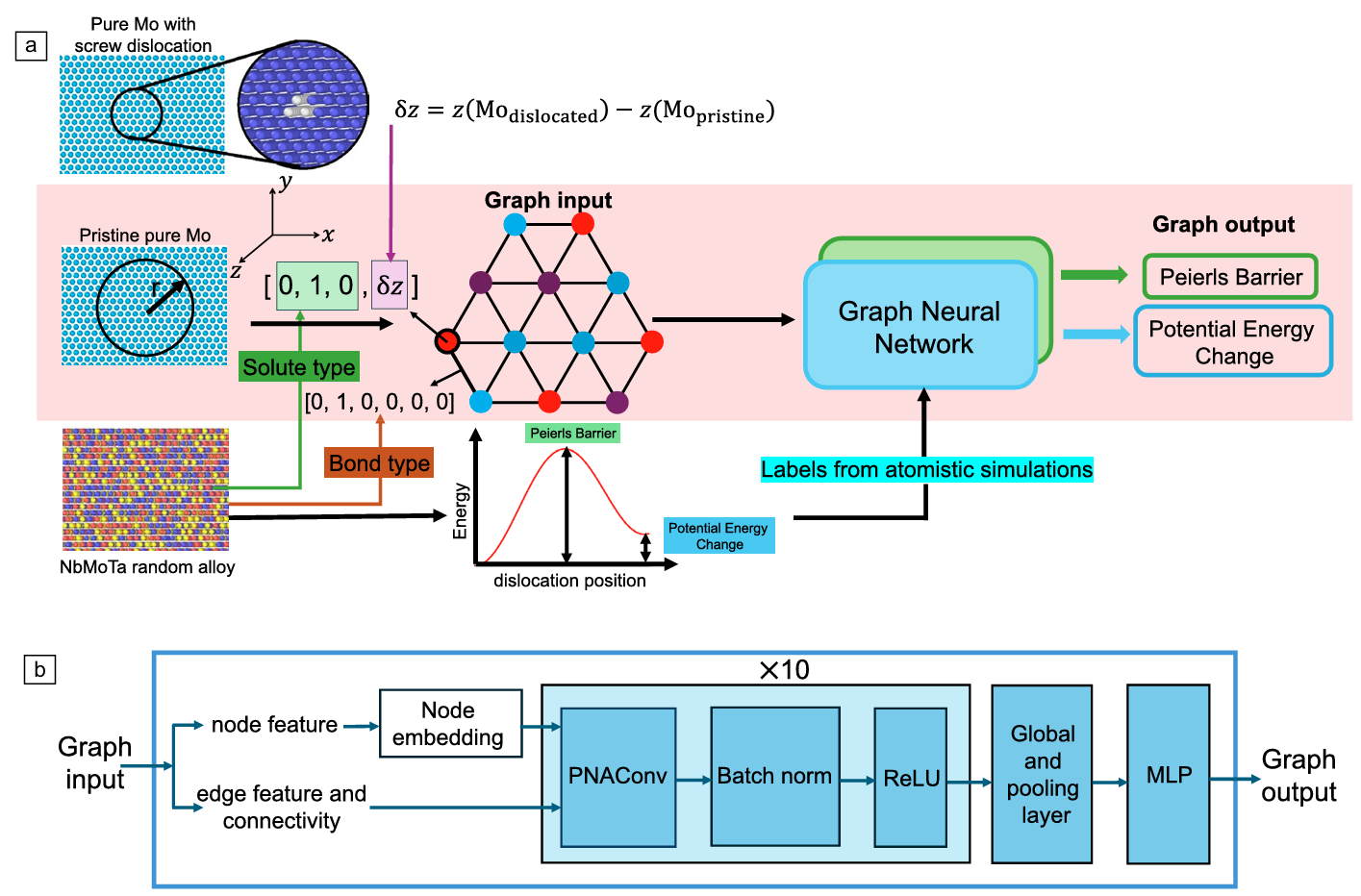
Figure 1. (a) Overview of the workflow used in this work to train graph neural network (GNN) models for an end-to-end prediction of the Peierls barrier and potential energy changes. (b) GNN architecture. Credit: Buehler et al.
Exploring Compositional Space and Predicting Performance
The experimental results demonstrate the system's capabilities across two primary use cases. In the first, the multi-agent system was tasked with exploring how the Peierls barrier varies across the entire niobium-molybdenum-tantalum ternary compositional space. Upon receiving the query, the assistant agent activated the planning tool, which through iterative collaboration between planner and reviewer agents, devised a comprehensive strategy. The plan specified generating 231 compositions at 5 percent intervals, computing Peierls barriers using the graph neural network, visualizing results on a ternary diagram, and conducting detailed analysis of trends and correlations.
Execution of this plan revealed fascinating insights into the compositional dependence of the Peierls barrier. The ternary plot shows regions of high barrier values around compositions with intermediate niobium and molybdenum and low tantalum content, while the lowest barriers appear in tantalum-rich compositions. The system then performed follow-up analysis on specific binary and ternary systems.
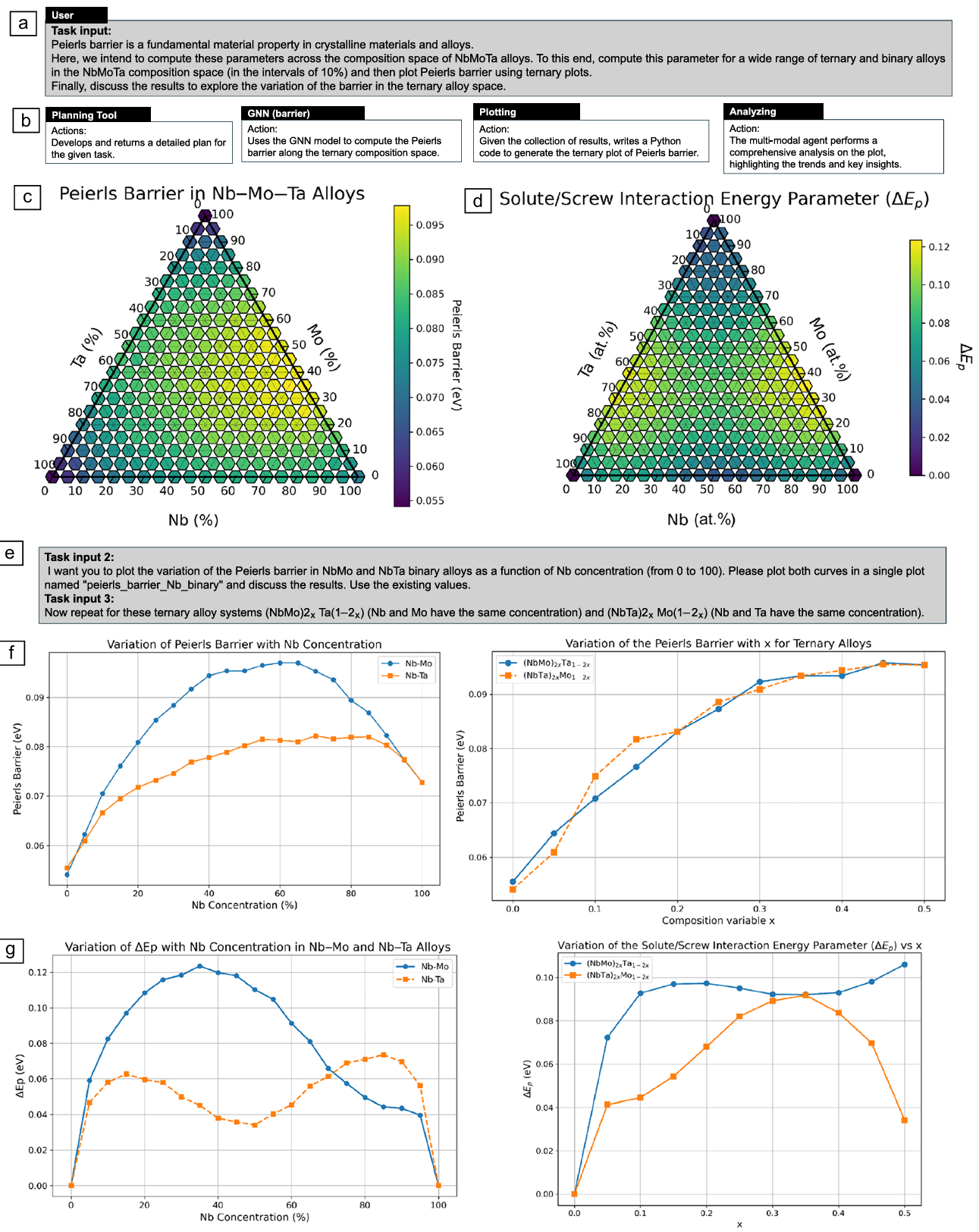
Figure 6. Overview of the multi-agent collaboration to explore the variation of the Peierls barrier and solute/screw interaction energy parameter in binary and ternary alloys. (a) User query requesting exploration of the Peierls barrier across a wide range of compositions, with a similar query repeated for the solute/screw interaction energy parameter. (b) Workflow detailing the computations and analyses performed by the multi-agent system. (c, d) Ternary plots displaying the Peierls barrier and the solute/screw interaction energy parameter, respectively, across the compositional space generated by the multi-agent system. (e) Follow-up tasks requested by the user, involving additional data plotting and analysis. (f, g) Plots illustrating the variation of the Peierls barrier and ˜Ep , respectively, with solute concentration for binary and ternary alloys, generated by the multi-agent model in response to the follow-up tasks. Credit: Buehler et al.
For the niobium-molybdenum binary system, the Peierls barrier increases with niobium concentration, peaking around 50 percent before decreasing as niobium content approaches 100 percent. The niobium-tantalum system shows a similar increasing trend but at a slower rate. These nonlinear variations reflect complex interactions between solute types and the screw dislocation core structure that would be difficult to predict from simple mixing rules.
The solute-screw interaction energy parameter, derived from potential energy changes as the dislocation moves between Peierls valleys, exhibits equally complex behavior. In the niobium-molybdenum system, this parameter increases with niobium concentration, peaking around 30 percent before gradually decreasing. In the niobium-tantalum system, higher values appear at high niobium concentrations compared to niobium-molybdenum.
For ternary systems like (NbTa)
The second major experiment tackled the more ambitious goal of predicting temperature-dependent yield stress for multiple alloy compositions. This required the multi-agent system to orchestrate a complex workflow including identifying the necessary input parameters, using the graph neural network to predict the Peierls barrier and solute-screw interaction energy, then estimating other parameters like lattice constant and kink formation energy by averaging over pure elements, feeding all inputs into the Maresca-Curtin theory implementation, generating predictions across a temperature range, and finally visualizing the results alongside experimental data for validation.
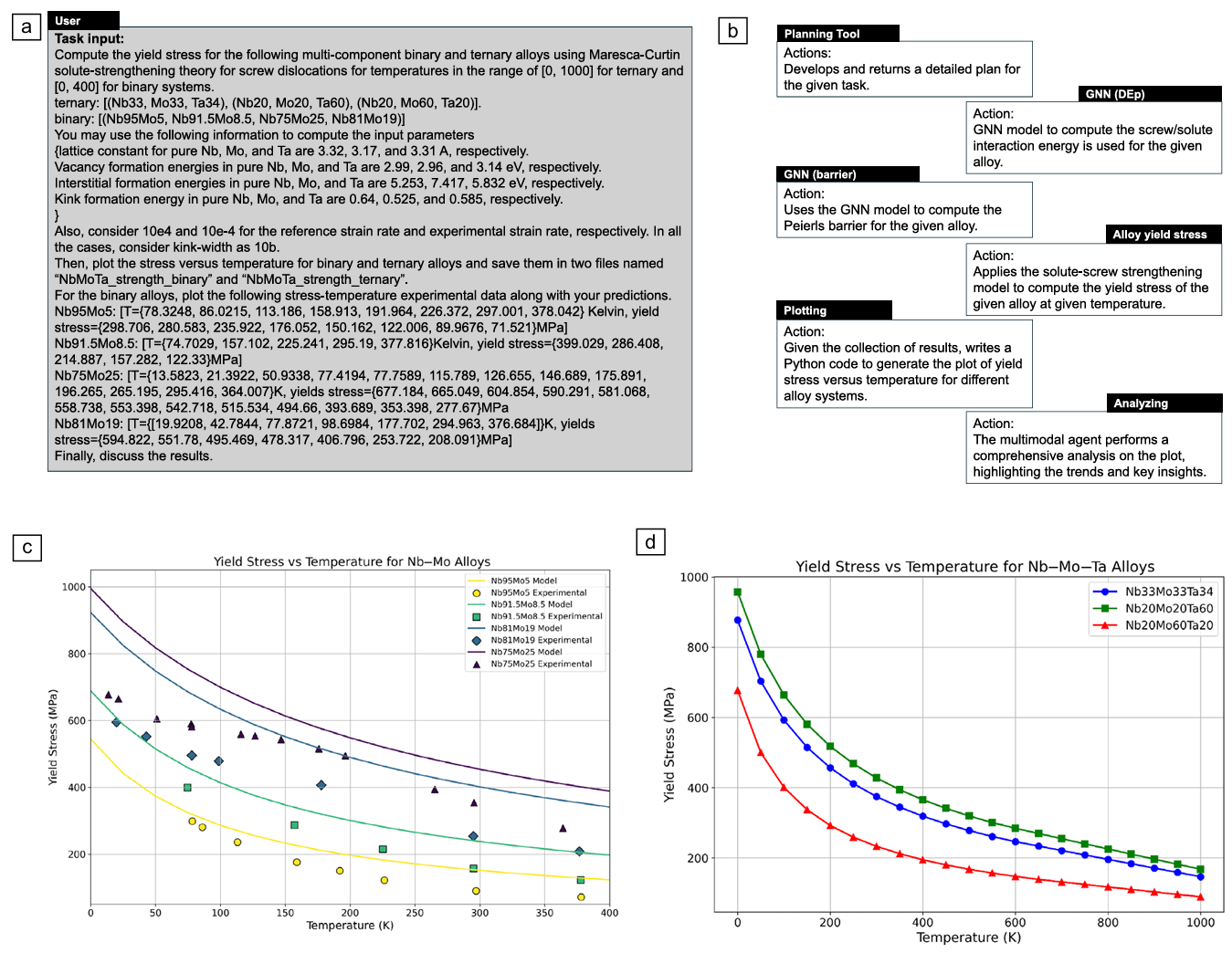
Figure 7. Overview of the multi-agent collaboration to predict the yield stress in binary and ternary alloy body-centered-cubic systems. (a) The input task involving computing the yield stress for a set of compositions. Some materials properties for the pure metals are pro vided as well as the strain rates. Moreover, the experimental data for binary alloys are provided to compare with the predictions. (b) The workflow of computations performed by our multi-agent system featuring key tasks such as planning, tool calling for material property predictions from the graph neural network (GNN) model, and finally plotting and analyzing the results. (c) Predictions of our model for the ternary alloys and (d) the predictions of our model for binary alloys along with the experimental results. Credit: Buehler et al.
The system executed this workflow seamlessly, producing plots showing yield stress versus temperature for both binary and ternary alloys. The results demonstrate the expected decrease in strength with increasing temperature, attributed to thermally activated processes that facilitate dislocation motion.
Comparison with experimental data revealed strong agreement for some compositions like Nb
While GPT-4o offered generic explanations focusing on composition and microstructure variations, the o1 model delivered more sophisticated physical insights, correctly attributing temperature effects to thermally activated kink-pair nucleation and propagation rather than diffusion-related atomic mobility.
These experiments illustrate several key advantages of the multi-agent approach. First, the graph neural network enables exploration of the full compositional space in a timeframe that would be impossible with traditional nudged elastic band calculations, which can require days per composition when accounting for statistical averaging over random configurations.
Second, the system demonstrates genuine reasoning capabilities, not just pattern matching. It can generate plans, execute complex workflows, handle unexpected results, and provide physical interpretations.
Third, the human-in-the-loop functionality allows users to ask follow-up questions, request different visualizations, or explore specific compositional regions in greater detail, making the system an interactive partner in the discovery process.
Lessons for the Future of Automated Discovery
The work by Ghafarollahi and Buehler represents a significant step toward fully automated materials discovery. By seamlessly integrating graph neural networks for rapid physics predictions, advanced large language models for planning and reasoning, specialized agents for different tasks, and established theoretical frameworks for connecting scales, the system demonstrates how artificial intelligence can accelerate the design cycle for complex materials.
The ability to explore 231 compositions in seconds, predict temperature-dependent mechanical properties, and provide physical interpretations of results showcases the potential for this approach to transform how researchers approach materials design challenges and work alongside AI Co-Scientist's.
Several aspects of the implementation deserve special recognition. The choice of graph neural network architecture, specifically the Principal Neighborhood Aggregation model, appears to have proved well-suited for capturing the relationship between atomic configurations and dislocation energetics.
The training strategy balanced data generation costs with model accuracy rather well. The multi-agent architecture's modular design, with distinct planning, coding, physics, and analysis tools, enables flexibility and extensibility. While the integration of advanced reasoning models like o1 for analysis tasks, shows how rapidly evolving large language model capabilities can enhance system performance.
Looking forward, the framework opens numerous research directions. The authors suggest integrating retrieval-augmented generation to access scholarly databases, incorporating domain-specific language models fine-tuned for materials science, and combining the multi-agent system with traditional optimization methods like genetic algorithms and active learning.
A particularly exciting possibility involves developing multi-agent models capable of autonomously formulating, refining, or improving physics-based theories themselves, moving beyond human-developed frameworks toward artificial intelligence-driven scientific discovery.
For researchers and practitioners in materials science and engineering, this work demonstrates the value of embracing multimodal approaches that combine the strengths of different artificial intelligence paradigms.
Graph neural networks excel at learning from structured data like atomic configurations. While large language models provide reasoning, planning, and natural language interaction capabilities. Specialized agents further enable task decomposition and parallel execution. Furthermore, physics-based theories ensure predictions remain grounded in fundamental principles. Together, these components create a system more powerful than any individual approach could achieve alone.
The availability of all data and code on GitHub at lamm-mit/AlloyAgents facilitates reproducibility and enables the community to build upon this work. The original AtomAgents framework is also available at lamm-mit/AtomAgents.
As artificial intelligence capabilities continue to advance, particularly in reasoning and multimodal understanding, systems like this will become increasingly sophisticated and capable.
The ultimate vision, where artificial intelligence collaborates with human researchers to accelerate discovery, optimize designs, and uncover new physical principles, moves closer to reality with each advancement. For the field of multi-principal element alloys and beyond, the future of materials discovery looks increasingly automated, intelligent, and promising.
Definitions
Body-Centered Cubic (BCC): A crystal structure where atoms are arranged at the corners of a cube with one atom at the center, characteristic of many refractory metals like niobium, molybdenum, and tantalum.
Graph Neural Network (GNN): A type of deep learning architecture designed to operate on graph-structured data, where nodes represent entities and edges represent relationships, making it ideal for modeling atomic structures.
Large Language Model (LLM): An artificial intelligence model trained on vast amounts of text data that can understand and generate human language, enabling capabilities like reasoning, planning, and code generation.
Multi-Principal Element Alloys (MPEAs): Alloys containing multiple elements in roughly equal proportions rather than one dominant element with minor additions, also known as high-entropy alloys.
Nudged Elastic Band (NEB): A computational method for finding minimum energy paths and transition states between initial and final atomic configurations, commonly used to calculate energy barriers for processes like dislocation motion.
Peierls Barrier: The intrinsic energy barrier that a dislocation must overcome to move through a perfect crystal lattice, representing the resistance to dislocation glide from the periodic potential of the crystal structure.
Principal Neighborhood Aggregation (PNA): A graph neural network architecture that combines multiple aggregation functions with degree-based scalers to improve performance on graph-level prediction tasks.
Screw Dislocation: A type of line defect in a crystal where the Burgers vector is parallel to the dislocation line, important for controlling plastic deformation in body-centered cubic metals.
Solute-Strengthening Theory: Theoretical frameworks that predict how dissolved atoms in an alloy interact with dislocations to impede their motion and increase material strength.
Yield Stress: The stress at which a material begins to deform plastically, representing the transition from elastic to plastic behavior and a critical property for structural applications.


Rapid and Automated Alloy Design with Graph Neural Network-Powered Large Language Model-Driven Multi-Agent AI
Rapid and automated alloy design with graph neural network-powered large language model-driven multi-agent AI