The escalating crisis of antibiotic resistance presents one of the most urgent threats to global public health, with millions of deaths linked to drug-resistant bacterial infections annually.
Among the most formidable is Staphylococcus aureus (S. aureus), particularly its methicillin-resistant strain (MRSA), which accounts for a significant portion of antimicrobial resistance (AMR)-related deaths worldwide. Despite this dire need, the development of new antibiotics has struggled to keep pace with the rapid evolution of resistance.
Generative Artificial Intelligence (AI) offers a beacon of hope in drug discovery, promising to accelerate the identification of novel therapeutic compounds. However, a significant hurdle for many existing AI models is their inability to consistently propose drug candidates that are both effective and synthetically tractable, meaning they can be realistically and efficiently manufactured in a laboratory setting. This disconnect between computational design and practical synthesis often limits the real-world impact of AI-driven drug discovery efforts.
To address these critical challenges, researchers at Stanford University introduce SyntheMol-RL, a reinforcement learning-based AI framework.
SyntheMol-RL is engineered to rapidly design novel small molecule drug candidates that are inherently synthetically accessible from a vast chemical space of 46 billion compounds. The technology represents a significant leap forward in bridging the gap between theoretical molecular design and practical drug development, particularly for urgently needed antibiotics.
Key Takeaways
- Generative AI shows immense promise for designing new drugs, but a major limitation of many existing models is their failure to propose compounds that are both effective and synthetically tractable.
- SyntheMol-RL significantly improves upon its Monte Carlo Tree Search (MCTS)-based predecessor, by generalizing across chemically similar building blocks and enabling multi-parameter optimization (e.g., antibacterial activity and aqueous solubility).
- The model is capable of exploring an enormous chemical space of 46 billion compounds, far exceeding typical virtual screening capabilities.
- SyntheMol-RL generated molecules with improved predicted properties and yielded a higher rate of potent in vitro hits compared to the previous MCTS version of SyntheMol and an AI-based virtual screening baseline.
- A significant achievement was the discovery of "synthecin". Synthecin demonstrated efficacy in a murine wound infection model of MRSA, validating the model's ability to generate real-world therapeutic candidates.
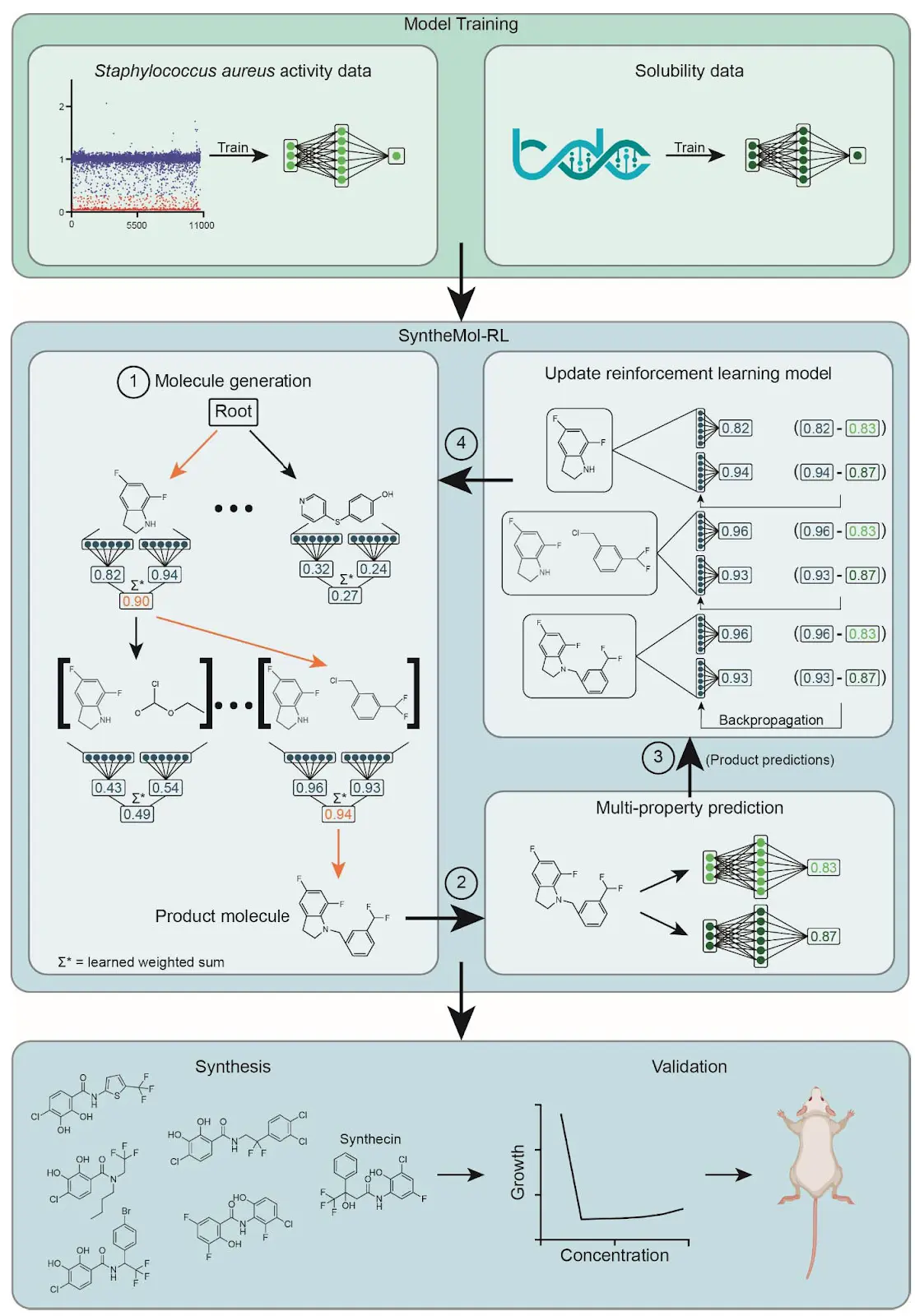
Figure 1. Overview of the SyntheMol-RL pipeline. The SyntheMol-RL pipeline can be broken into three primary steps. (1) Property prediction models are trained based on a combination of in-house chemical screening data against S. aureus as well as aqueous solubility data obtained from the Therapeutic Data Commons. (2) SyntheMol-RL generates molecules guided by these property prediction models. A reinforcement learning (RL) model is used to iteratively select molecular building blocks based on their potential for generating molecules with promising S. aureus antibacterial activity and aqueous solubility (RL scores shown in blue). The generated molecule is then scored using the property prediction models, and these scores are used to update the RL model to improve its generative capacity (property prediction scores shown in green). (3) A prioritized subset of molecules is selected for synthesis and laboratory testing based on predicted properties as well as novelty and diversity. Notably, this SyntheMol-RL pipeline generated a novel compound that can effectively treat a MRSA wound infection in a mouse model, showing the utility of SyntheMol-RL for real-world drug discovery applications.
Overview
The global health landscape is increasingly threatened by antimicrobial resistance (AMR), where microorganisms, such as bacteria, viruses, fungi, and parasites, evolve to withstand the effects of antimicrobial drugs. This resistance renders infections harder to treat, increasing the risk of disease spread, severe illness, and death.
In 2019 alone, approximately 4.95 million deaths were linked to drug-resistant bacterial infections, a figure projected to rise to 10 million annually by 2050 if the current trend continues.
Among the most concerning threats are the ESKAPE pathogens,a group of six bacterial species (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) notorious for their virulence and multi-drug resistance.
Staphylococcus aureus, particularly its methicillin-resistant variant (MRSA), stands out as a leading cause of morbidity and mortality, responsible for a quarter of AMR-associated deaths in high-income regions. The World Health Organization (WHO) has highlighted S. aureus as a high priority for new antibiotic development due to the diminishing treatment options.
In response to this urgent need, Artificial Intelligence (AI) has emerged as a transformative force in drug development. A common AI approach involves property prediction models, which are trained to forecast molecular characteristics like antibacterial activity or aqueous solubility.
These models can then be used in virtual screenings, where large chemical libraries are evaluated to identify promising compounds. However, this method is computationally intensive and doesn't scale well to the massive chemical spaces, often exceeding billions of molecules, now available from commercial databases like Enamine’s REAL Space and WuXi’s GalaXi. To overcome the slow, molecule-by-molecule evaluation of virtual screening, generative AI methods have been developed to directly design molecules with desired properties.
While promising, a significant limitation of many generative AI models has been the synthesizability of the proposed compounds. Often, AI-generated molecules, despite favorable predicted efficacy, prove synthetically intractable, hindering experimental validation and real-world application.
To tackle this, previous research introduced SyntheMol, a generative AI model that employed a Monte Carlo Tree Search (MCTS) algorithm. The MCTS version treated the molecule generation process as a tree search, where it iteratively selects molecular building blocks and combines them using predefined chemical reactions to form molecules.
While SyntheMol successfully designed compounds with in vitro antibacterial activity against Acinetobacter baumannii and other ESKAPE species, it faced two key limitations:
- Inefficient Chemical Space Exploration: MCTS treats each building block independently, failing to learn and generalize patterns of antibacterial activity across chemically similar blocks limiting its search efficiency within large chemical spaces.
- Single-Parameter Optimization: The original SyntheMol could only optimize one molecular property at a time. Real-world drug discovery, however, necessitates optimizing multiple druglike properties simultaneously (e.g., activity, solubility, and toxicity). For instance, prior work found that only two of six potent antibacterial compounds generated by SyntheMol were sufficiently soluble for in vivo (animal model) experiments.
SyntheMol-RL replaces the MCTS algorithm with a reinforcement learning (RL) model. In RL, a deep learning model acts as a value function, learning to predict the expected property score of molecules based on the chemical structures of the building blocks chosen. This allows SyntheMol-RL to generalize across chemically similar building blocks, leading to more rapid and effective exploration of combinatorial chemical spaces.
The new version also introduces multi-parameter optimization, enabling the generation of compounds that simultaneously possess multiple desired molecular properties, such as antibacterial activity and aqueous solubility.
The model dynamically adjusts the weights given to each property over time to explore the Pareto frontier (the Pareto frontier represents the set of optimal solutions in a multi-objective optimization problem showing the best possible trade-offs between different, often conflicting, objectives), identifying areas with the highest density of "hit" molecules (those meeting user-defined thresholds for all properties).
This new framework also expands the chemical space explored from 30 billion to an even larger 46 billion compounds, drawing from the Enamine REAL Space and WuXi GalaXi databases.
Why it's important
SyntheMol-RL's tackles a critical bottleneck in AI-driven drug discovery: the gap between computationally designed molecules and their real-world synthesizability and efficacy.
The ability to rapidly design compounds that are both effective and easily synthesizable by design is potentially revolutionary for the pharmaceutical industry and public health. SyntheMol-RL intrinsically addresses this by constraining its generative process to combinatorial chemical spaces built from readily available building blocks and well-validated chemical reactions.
The importance of SyntheMol-RL is highlighted through its application to antibiotic discovery. The discovery of "synthecin," a structurally novel compound effective against MRSA in a mouse model, is a direct validation of SyntheMol-RL's capacity to deliver urgently needed new antibiotics.
This is not just a theoretical finding; it's a demonstration of a pathway to new drugs that can save lives. The fact that SyntheMol-RL identified molecules with a higher hit rate (19% of synthesized SyntheMol-RL compounds were potent in vitro hits) compared to traditional virtual screening or previous generative models underscores its efficiency and potential to accelerate the discovery pipeline.
The "design-for-synthesis" approach ensures that the output is not just theoretically optimal but also experimentally achievable, significantly shortening the time and cost associated with drug development. The direct validation of 79 synthesized compounds, with 13 showing potent in vitro activity and 7 being structurally novel, suggests the practical relevance of SyntheMol.
Beyond antibiotics, SyntheMol-RL's underlying methodology offers broad implications for the entire drug discovery field:
- Multi-parameter Optimization: The ability to simultaneously optimize for multiple drug-like properties (e.g., activity, solubility, ADMET properties) is crucial for developing viable drug candidates. SyntheMol-RL's dynamic property weight tuning adjusts the focus between different objectives to maximize hits. It is a sophisticated approach that could be adapted for various drug classes and targets or even other scientific disciplines.
- Efficient Exploration of Massive Spaces: By leveraging reinforcement learning to generalize across chemical similarities, SyntheMol-RL can navigate chemical spaces of tens of billions of molecules more efficiently than traditional methods. This is vital as industrial and academic settings increasingly utilize ultra-large databases.
- Facilitating Structure-Activity Relationship (SAR) Studies: The model's tendency to generate clusters of molecules around a shared building block, while diversifying secondary building blocks, is an inherently valuable characteristic for medicinal chemistry. This clustering allows for the natural exploration of the SAR landscape, making it easier to identify molecular substructures responsible for activity and to optimize compounds further.
- Flexibility and Adaptability: SyntheMol-RL's compatibility with any property predictor and combinatorial chemical space makes it a highly flexible tool. This means it can be readily extended to design compounds for a wide array of diseases and molecular targets beyond bacterial infections, from anti-cancer agents to neuroscience drugs.
In essence, SyntheMol-RL represents a powerful computational engine that can transform the early stages of drug discovery by intelligently designing molecules that are not only predicted to be effective but are also ready for immediate experimental validation, helping to accelerate the pipeline from idea to life-saving medications.
Summary of Results
The research details a systematic approach to developing and validating SyntheMol-RL for the discovery of novel and synthesizable antibiotics, focusing on Staphylococcus aureus. The framework is open-source and available on Github.
The researchers were able to train each of the four model-dataset combinations (Chemprop-RDKit and MLP-RDKit on both antibacterial activity and aqueous solubility datasets) trained in less than 80 minutes on consumer hardware (8CPU & 1 GPU). Molecule generation took up to 7 days while virtual screening took roughly 7 days on the same hardware.
The ability to run the application on consumer grade hardware democratizes the availability of cutting edge chemistry tools to individuals and small labs helping to further scientific discovery.
Property Prediction Model Development
This step involved training robust molecular property prediction models to guide SyntheMol-RL.
The focus was on two key properties:
- Antibacterial Activity: Defined as the ability to inhibit S. aureus RN4220 growth in vitro. A custom training set of 10,716 compounds was physically screened, yielding 1,137 active and 9,521 inactive unique molecules.
- Aqueous Solubility: Essential for in vivo testing and drug delivery, this data was sourced from the AqSolDB dataset (9,982 molecules). This was chosen to address prior solubility issues encountered with SyntheMol-MCTS.
Two model architectures were employed for property prediction:
- Chemprop-RDKit: A Graph Neural Network (GNN) model (Chemprop) augmented with 200 molecular features from RDKit. This model learns comprehensive molecular representations.
- MLP-RDKit: A Multilayer Perceptron (MLP) that only uses the 200 RDKit features as input, without the GNN component. This model is faster but potentially less accurate.
Both models were trained using 10-fold cross-validation. Chemprop-RDKit consistently showed slightly higher performance than MLP-RDKit across both properties [Figure 2d].
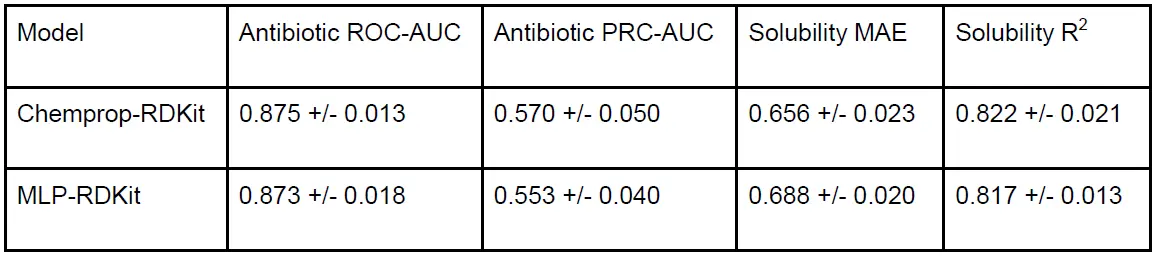
Table 1. Property Prediction Model Performance The table above shows the performance metrics (ROC-AUC, PRC-AUC for antibiotic activity; MAE, R2 for solubility) for the two property prediction model architectures. Both models demonstrated high performance, with Chemprop-RDKit slightly outperforming MLP-RDKit.
Generative Model Development (SyntheMol-RL vs. SyntheMol-MCTS)
The original SyntheMol used Monte Carlo Tree Search (MCTS) to select building blocks and reactions within a combinatorial chemical space. MCTS calculates a "value" for each node (combination of building blocks) based on how often it's explored and the average property score of molecules built from it.
However, MCTS treats building blocks independently, failing to learn chemical patterns, and could only optimize one property at a time.
SyntheMol-RL overcomes these limitations by:
- Reinforcement Learning (RL): Replaced MCTS with an RL algorithm where a deep learning model serves as a value function. This value function learns to predict the expected property score of molecules from their building blocks, enabling generalization across chemically similar blocks.
- Multi-parameter Optimization: The value of a node in SyntheMol-RL is a weighted combination of multiple RL models, one for each property (e.g., antibacterial activity and aqueous solubility). The model dynamically adjusts these weights to find the Pareto frontier of optimal multi-property compounds, maximizing the discovery of "hit" molecules.
- Expanded Chemical Space: SyntheMol-RL explores 46 billion compounds from two major combinatorial chemical spaces: the Enamine REAL Space (31 billion molecules from ~139,000 building blocks and 169 reactions) and the WuXi GalaXi (16 billion molecules from ~15,000 building blocks and 36 reactions).
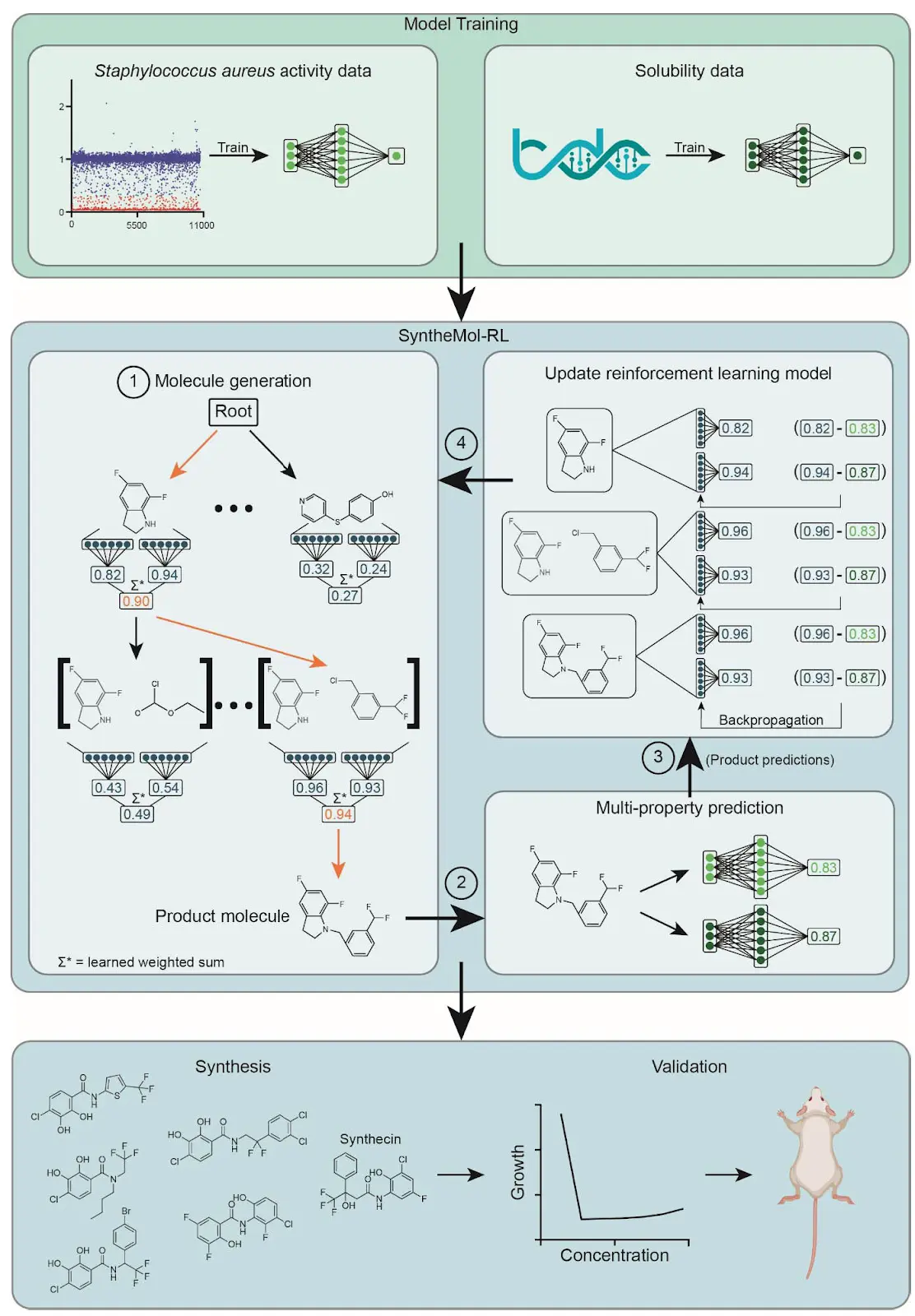
Figure 1. Overview of the SyntheMol-RL pipeline The diagram illustrates the three primary steps: (1) Training property prediction models using S. aureus screening and solubility data. (2) SyntheMol-RL generates molecules guided by these models, using an RL model to iteratively select building blocks based on their potential for desired properties (RL scores in blue). Generated molecules are scored by property prediction models (green), and these scores update the RL model. (3) A prioritized subset of molecules is selected for synthesis and testing based on predicted properties, novelty, and diversity. The overall pipeline led to the discovery of a novel compound effective in a MRSA wound infection model.
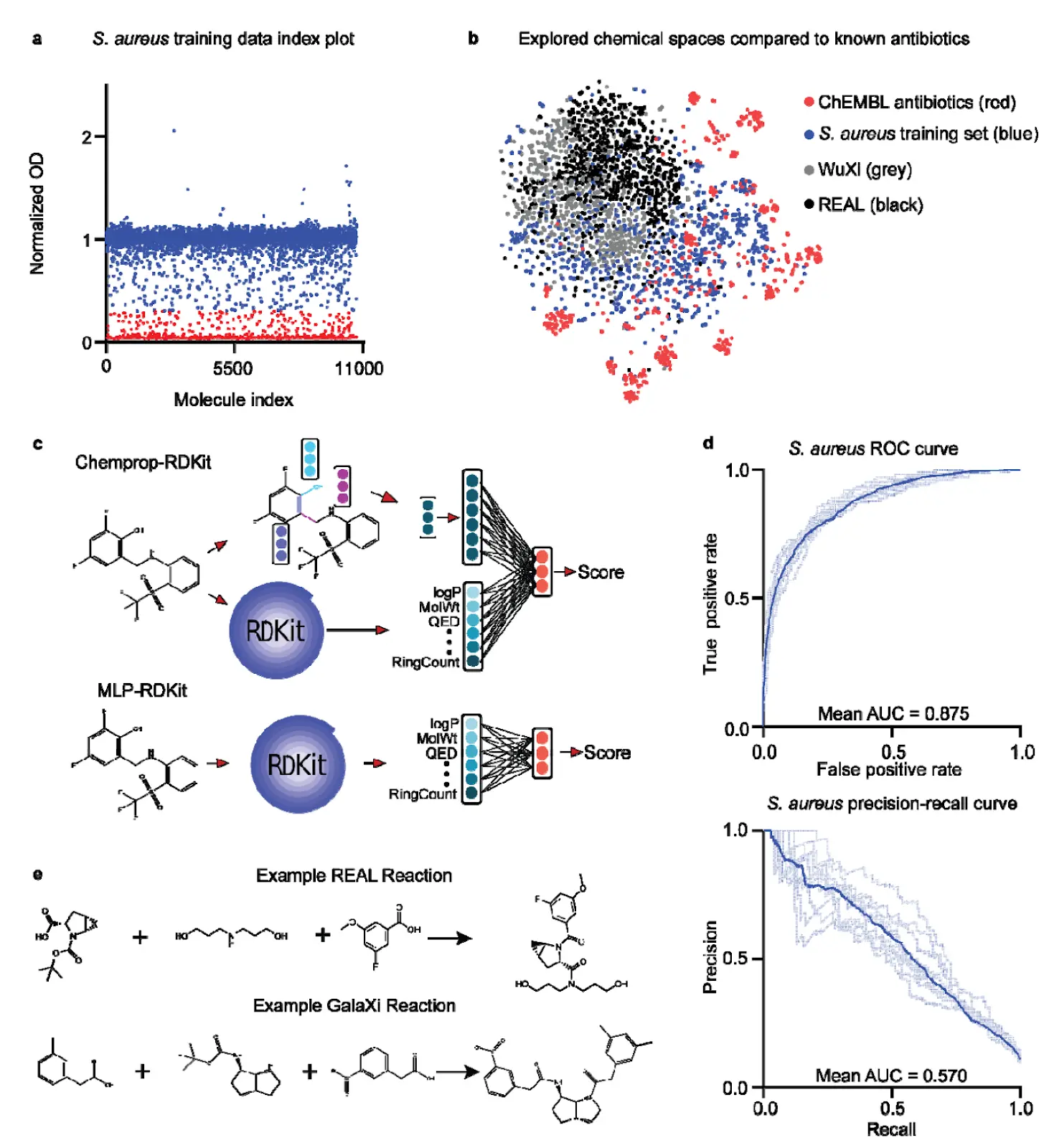
Figure 2. Property prediction models and chemical spaces Panel a shows the distribution of S. aureus growth inhibition values from the screening of 10,716 compounds. Red points are "hits" (below threshold), blue are "non-hits." Panel b provides a t-SNE visualization comparing the chemical spaces of ChEMBL antibiotics, the S. aureus training set, and the REAL and WuXi GalaXi spaces explored by SyntheMol-RL, demonstrating chemical diversity. Panel c depicts the Chemprop-RDKit (top) and MLP-RDKit (bottom) model architectures. Panel d shows the ROC and precision-recall curves for the S. aureus activity property prediction models. Panel e provides example reactions from the Enamine REAL Space and WuXi GalaXi, illustrating how molecules are formed from building blocks.
Generative Model Application
SyntheMol-RL was applied to design antibiotics against S. aureus, with simultaneous optimization for antibacterial activity and aqueous solubility. Two versions were run:
- RL-Chemprop: Used Chemprop-RDKit as the RL value function architecture.
- RL-MLP: Used MLP-RDKit as the RL value function architecture.
For comparison, the original MCTS model and a Virtual Screening approach (VS-Chemprop) were also run. VS-Chemprop evaluated 21 million randomly sampled molecules (14M from REAL, 7M from GalaXi) to match the computational time of the slowest generative model.
The results demonstrated SyntheMol-RL's superior performance in silico:
- Hit Rates: Defining a "hit" as antibacterial prediction score ≥ 0.5 and log solubility ≥ -4, RL-Chemprop generated 11.6% hits (1,273 of 10,983 molecules), and RL-MLP generated 5.3% hits (493 of 9,228). This significantly outperformed MCTS (3.0% hits) and VS-Chemprop (0.006% hits).
- Antibacterial Activity: Generative models, especially RL models, excelled at designing molecules with high predicted antibacterial activity, while all methods could identify soluble molecules [Figure 3b, 3c].
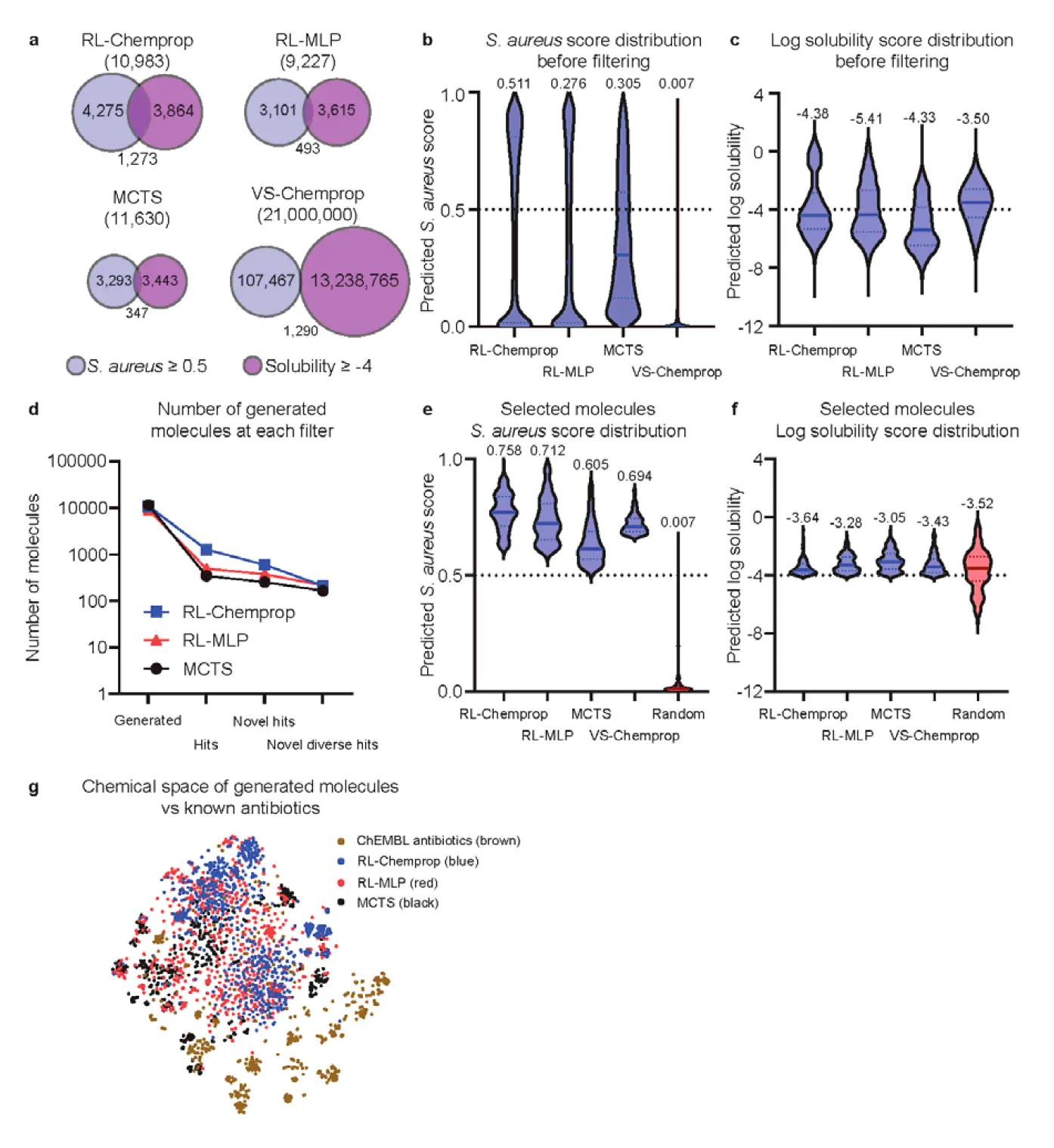
Figure 3. Generating compounds with SyntheMol-RL Panel a uses Venn diagrams to show the number of molecules from each method (RL-Chemprop, RL-MLP, MCTS, VS-Chemprop) that meet predicted S. aureus activity and solubility thresholds, highlighting the superior hit rates of RL models. Panels b and c display violin plots of predicted S. aureus scores and log solubility scores respectively, across the methods, showing that generative models (especially RL) are better at designing high-activity molecules. Panel d illustrates the filtering process for selecting compounds for synthesis. Panels e and f show the distributions of predicted S. aureus and log solubility scores for the filtered sets, including a random baseline, reaffirming RL-Chemprop's aptitude for generating likely antibiotic candidates. Panel g provides a t-SNE visualization of the training set, generated molecules, and known antibiotics, demonstrating the diversity and novelty of generated compounds.
To select promising compounds for synthesis and in vitro testing, a rigorous filtering process was applied:
- Predicted Hit: Antibacterial score ≥ 0.5 and log solubility ≥ -4.
- Novelty: Maximum Tversky similarity ≤ 0.6 compared to known antibiotics.
- Diversity: Tanimoto similarity ≤ 0.6 between any two compounds in the selected set.
- Top Ranked: Top 150 compounds by antibacterial score from each method (RL-Chemprop, RL-MLP, MCTS, VS-Chemprop) were selected, plus 150 random compounds, totaling 750 unique compounds.
Further filters for availability from vendors (Enamine and WuXi) and lowest predicted clinical toxicity (using ADMET-AI) narrowed the selection to 250 compounds (50 per method type), of which 194 unique compounds were successfully synthesized for experimental validation. Notably, SyntheMol-RL (and MCTS) showed a strong preference for Enamine compounds over WuXi, likely due to Enamine's larger building block selection.
A computational benchmark against GFlowNet, a state-of-the-art generative model, showed that while GFlowNet produced molecules with higher predicted scores, they were often bulkier and structurally complex, leading to poor whole-cell activity and significantly higher synthesis costs (>30x). This highlighted SyntheMol-RL's advantage in generating practical, synthesizable candidates.
In Vitro Validation of AI-Generated Molecules
The 194 synthesized compounds were tested for antibacterial activity against S. aureus RN4220 using Minimum Inhibitory Concentration (MIC) assays.
- Hit Rate: 11 of 38 (29%) compounds from RL-Chemprop and 2 of 41 (5%) from RL-MLP showed potent activity (MIC ≤ 8 µg/ml) [49, Figure 4a]. In contrast, the 44 randomly selected molecules showed no activity. MCTS and VS-Chemprop yielded 0 and 2 hits, respectively.
- Structural Novelty: Of the 13 identified hits, a thorough manual literature search confirmed seven to be structurally novel, with six from RL-Chemprop and one from RL-MLP [Figure 4b]. The other hits had structural overlap with known antibacterial classes like salicylanilides.
- Spectrum Activity: The seven novel compounds were tested against a diverse panel of ESKAPE pathogens. They showed no significant potency against other Gram-positive or Gram-negative bacteria, indicating a narrow-spectrum activity primarily against S. aureus [Figure 4c]. This is a beneficial characteristic for minimizing disruption to host microbiota.
- Activity Against Resistant Strains: Importantly, these seven novel compounds retained potent activity (MIC ≤ 8 µg/ml) against clinically relevant antibiotic-resistant S. aureus strains, including MRSA USA300 and multidrug-resistant vancomycin-intermediate S. aureus (VISA) isolates covering all known resistance mechanisms [Figure 4c]. This strongly suggests their ability to overcome common resistance determinants due to their structural novelty.
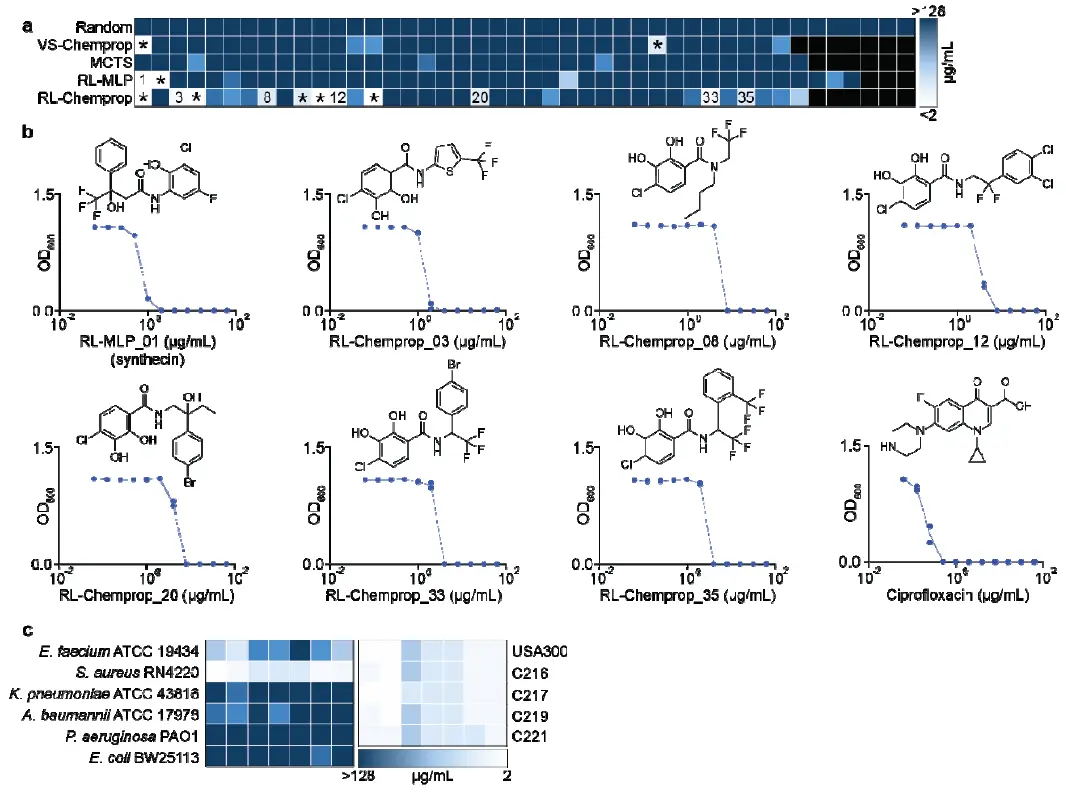
Figure 4. Generated molecules show potent in vitro activity Panel a presents a heatmap of MICs against S. aureus RN4220 for compounds from each method, showing the high hit rates of RL-Chemprop and RL-MLP. Panel b shows the growth inhibition curves and structures of the seven identified structurally novel molecules. Panel c displays heatmaps of MICs for the seven novel compounds against ESKAPE pathogens (left) and various antibiotic-resistant S. aureus clinical isolates (right), demonstrating their narrow-spectrum activity but robust potency against resistant S. aureus strains.
In Vivo Validation of SyntheMol-RL-Generated Molecules
One particularly potent and structurally novel compound, MLP-01, was named synthecin and selected for further in vivo characterization.
- C.R.E.A.M. Assay: A Chemical Release Evaluation on Agar Media (C.R.E.A.M.) assay was developed as an in vitro model mimicking in vivo topical application. Synthecin, formulated in Glaxal Base, showed the largest zone of growth inhibition, indicating promising topical antibacterial efficacy [Figure 5a]. All seven novel compounds were readily soluble in Glaxal Base, a confirmation of the solubility optimization during generation.
- Bacteriostatic Activity: Synthecin demonstrated bacteriostatic activity against S. aureus USA300 in liquid media, similar to linezolid, a commonly prescribed antibiotic for S. aureus skin infections [Figure 5b].
- Murine Wound Infection Model: Synthecin's efficacy was tested in a mouse wound infection model using MRSA USA300. Mice treated topically with synthecin showed a significantly lower bacterial burden in their wounds (~5.14 × 103 CFU/g) compared to vehicle-treated mice (~6.39 × 107 CFU/g) [Figure 5d]. Synthecin-treated tissues displayed no signs of inflammation, unlike the vehicle controls [Figure 5e].
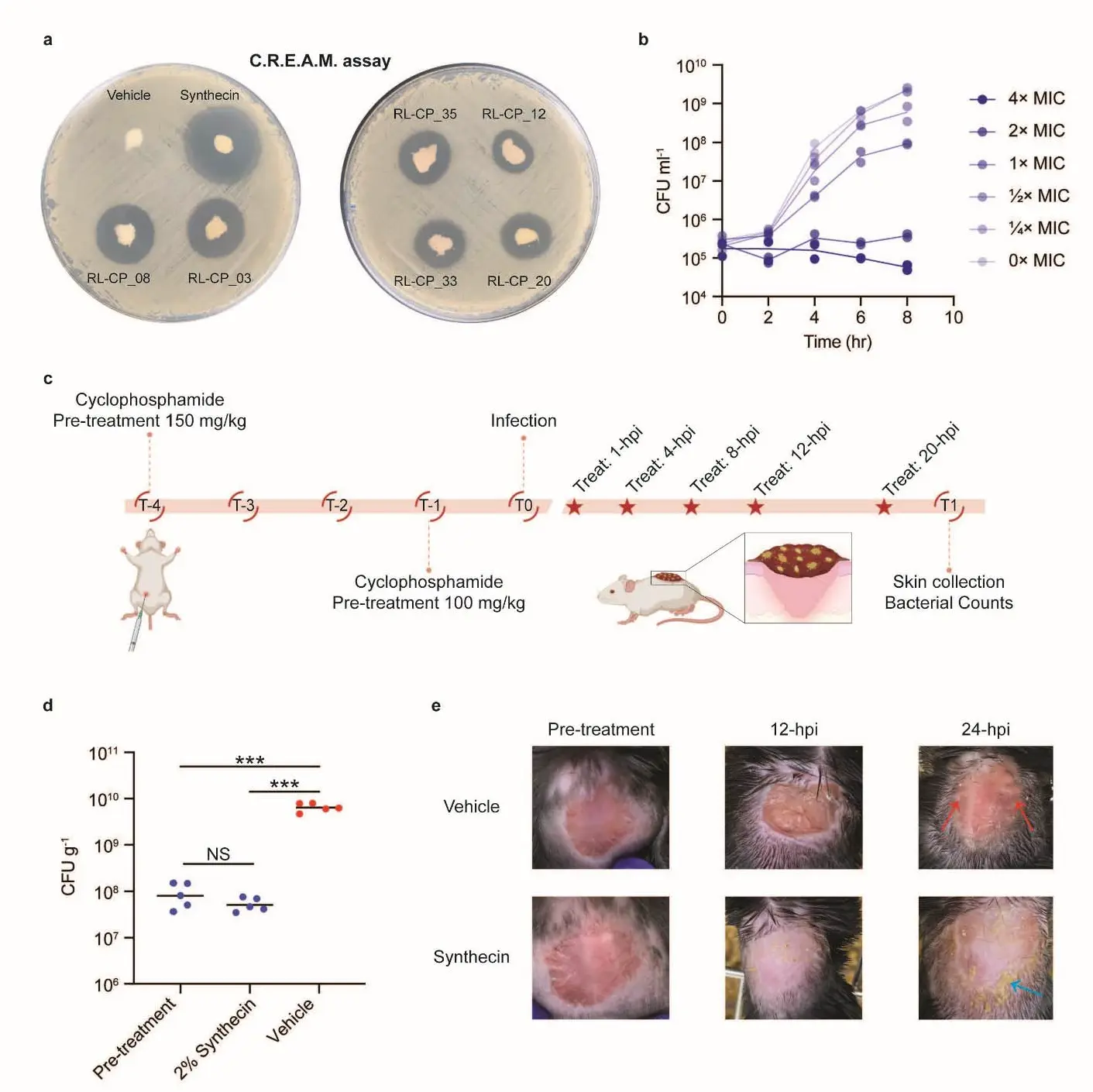
Figure 5. Synthecin can suppress an S. aureus infection in a mouse wound model Panel a shows the C.R.E.A.M. assay results, where synthecin (MLP-01) produced the largest zone of inhibition, correlating with its in vivo efficacy. Panel b illustrates synthecin's bacteriostatic activity against S. aureus USA300. Panel c outlines the experimental design for the murine wound infection model. Panel d presents the bacterial load in wound tissue, showing a significant reduction in MRSA burden in synthecin-treated mice compared to controls. Panel e provides representative images of mouse dorsal wounds, visually confirming reduced inflammation and bacterial load in synthecin-treated mice.
These results collectively validate SyntheMol-RL's ability to generate novel and synthetically accessible antibiotic candidates, culminating in the successful in vivo demonstration of synthecin's potent activity against a prevalent and drug-resistant bacterial infection.
Conclusion
SyntheMol-RL stands as a testament to the transformative potential of artificial intelligence in addressing some of humanity's most pressing health challenges. By marrying the power of reinforcement learning with a principled "design-for-synthesis" approach, this generative AI model has demonstrated an unprecedented ability to rapidly explore vast chemical spaces and identify novel, effective, and synthetically accessible drug candidates.
The successful application to the urgent global crisis of antibiotic resistance is particularly compelling. SyntheMol-RL not only generated a superior set of S. aureus antibiotic candidates based on in silico predictions and in vitro experiments compared to previous models and virtual screening, but it also directly led to the discovery and validation of synthecin.
The finding that synthecin, a structurally novel molecule, can effectively suppress MRSA wound infections in a mouse model bridges the critical gap between computational design and real-world therapeutic impact. This highlights SyntheMol-RL's utility not just as a research tool but as a flexible framework for readily translatable drug discovery.
The innovations embedded within SyntheMol-RL, such as its capacity for multi-parameter optimization and dynamic parameter tuning, represent significant advancements in molecular design. Its inherent ability to bias generation towards synthesizable compounds, in contrast to many other generative models that struggle with synthetic feasibility, makes it a highly practical tool for pharmaceutical research and development.
Moreover, the tendency of SyntheMol-RL to generate compounds in chemically intuitive clusters offers a natural advantage for Structure-Activity Relationship (SAR) studies, allowing researchers to efficiently optimize promising scaffolds.
Looking forward, the flexibility of SyntheMol-RL, being compatible with any property predictor and combinatorial chemical space, means its potential extends far beyond antibiotics. It can be readily adapted to discover drugs for a wide array of therapeutic domains, from oncology to neurological disorders, by simply changing the target properties and chemical building blocks.
Further refinements to the RL algorithm itself and exploration into optimizing an even larger number of objectives simultaneously could unlock even greater drug-like characteristics in generated compounds. Ultimately, SyntheMol-RL embodies a powerful paradigm for accelerating drug discovery, ensuring that the promise of AI translates into tangible, life-saving medicines that can be brought to patients more rapidly and efficiently than ever before.


Introducing SyntheMol-RL An Antibiotic Discovery Powered by Artificial Intelligence
SyntheMol-RL: a flexible reinforcement learning framework for designing novel and synthesizable antibiotics