Protein folding is the process of predicting a protein's three-dimensional atomic structure from its amino acid sequence which has been revolutionized by deep learning breakthroughs like AlphaFold2 and RoseTTAFold (Jumper et al., 2021; Baek et al., 2021).
These sophisticated systems leverage carefully engineered domain-specific components: multiple sequence alignments (MSA), pair representations, and triangle updates. While they achieve remarkable accuracy on community benchmarks, they raise a fundamental question: what if we could match their performance using simpler, more general-purpose architectures?
A compelling new preprint paper from Apple Research challenges this complexity. The authors introduce a transformer-only, flow-matching folding model that deliberately abandons MSA processing, pair representations, and triangular updates.
In their place, SimpleFold relies on standard transformer blocks with adaptive layers and a generative flow-matching training objective. By scaling to 3B parameters and training on millions of distilled structures plus Protein Data Ban (PDB) data, they achieve competitive accuracy on CAMEO22 and CASP14 while excelling at ensemble generation, all with practical inference speeds on consumer hardware.
We analyzed the complete paper, examining every figure and table, and verified all key references against their original sources.
Key Takeaways
- Transformer-only architecture: SimpleFold eliminates MSA processing, pair representations, and triangle updates, using a flow-matching objective to generate full-atom structures directly from sequence.
- Competitive accuracy: On CASP14, the 3B parameter SimpleFold achieves TM-score 0.720/0.792 and GDT-TS 0.639/0.703 versus ESMFold's 0.701/0.792 and 0.622/0.711 (Table 1).
- Superior ensemble generation: Without extra tuning, SimpleFold better matches molecular dynamics (MD) distributions than MSA-based baselines. A fine-tuned variant (SimpleFold-MD) shows further improvements (Table 2).
- Scaling benefits: Larger models particularly excel on challenging CASP14 targets, with Figure 4 demonstrating clear positive scaling trends.
- Practical efficiency: The smallest 100M parameter model achieves ~90% of the larger model's CAMEO22 performance while running efficiently on consumer-grade hardware (Figure 1e).
Overview
SimpleFold reframes protein folding as conditional generative modeling where given an amino acid sequence, the model generates full-atom coordinates by integrating a learned, time-dependent vector field that transforms noise into structured data.
This approach follows the flow matching paradigm (Lipman et al., 2022; Albergo and Vanden‑Eijnden, 2023), which regresses the model's velocity to a target velocity along simple paths such as a linear interpolant.
Concretely, the paper uses the interpolantwith target velocity , training a network via an loss augmented with a local LDDT term. At inference, the model integrates the learned field from to , optionally using a Langevin‑style SDE formulation (Euler–Maruyama), to stochastically sample plausible conformations.
Architecturally, SimpleFold uses three components built from standard transformer blocks with adaptive layers (time conditioning): a light atom encoder, a heavy residue "trunk," and an atom decoder (Figure 2).
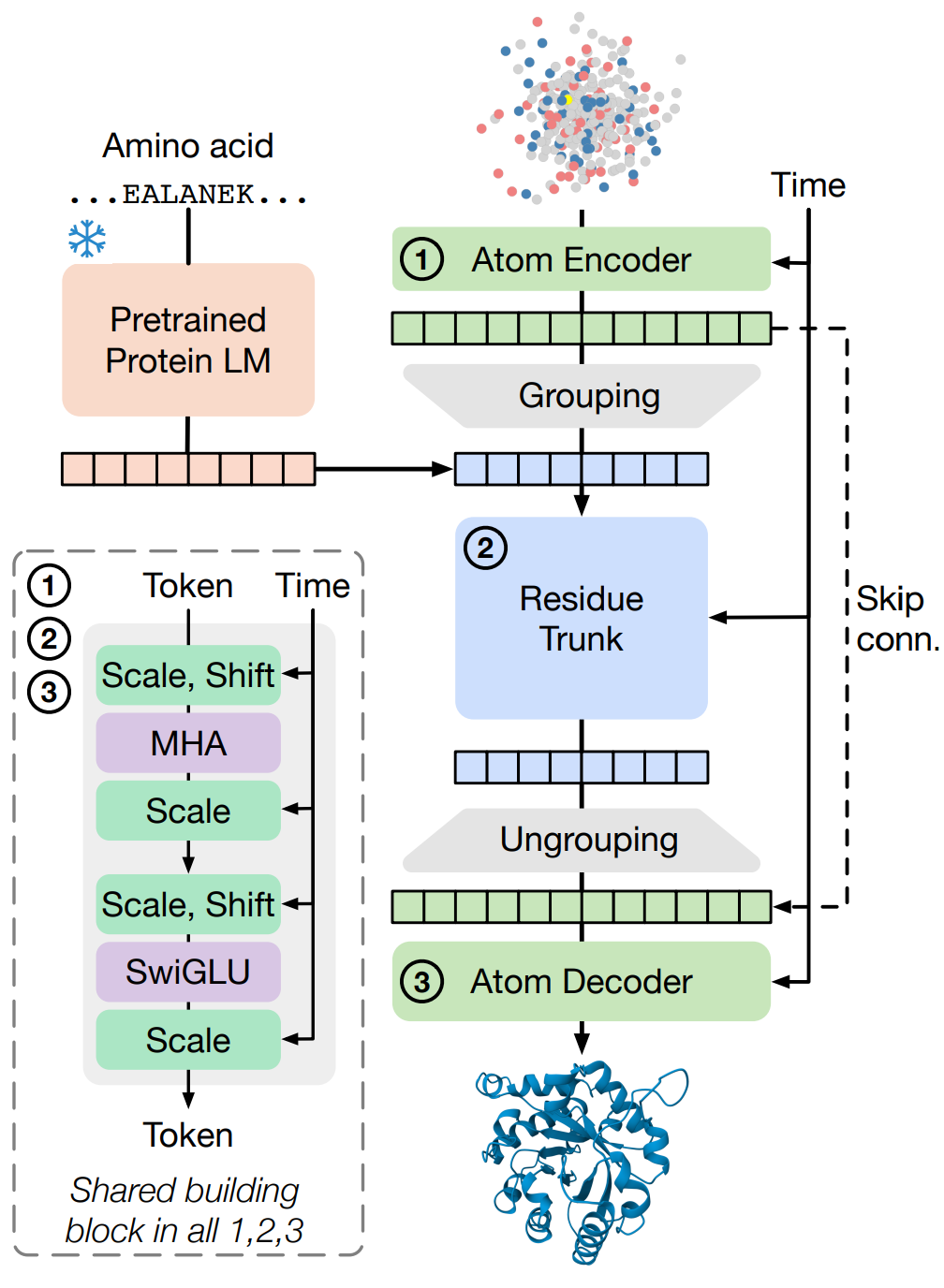
Figure 2 Overview of SimpleFold’s architecture built on general-purpose standard Transformer block with adaptive layers. Atom encoder, residue trunk, and atom decoder all share the same general-purposed building block. Our model circumvents the need for pair representations or triangular updates.
The encoder operates with local attention masks over atoms, then "groups" to per‑residue tokens; while the trunk processes residue tokens (concatenated with frozen ESM2‑3B sequence embeddings) and the decoder "ungroups" back to atoms and predicts the velocity field.
Rotary position embeddings (RoPE) are used for sequence positions and a 4D axial variant encodes local structure context for atoms. The model is intentionally non‑equivariant, instead of equivariant layers, the authors rely on data augmentation (with random SO(3) rotations) and capacity to learn symmetry.
Data scale is central and for most models, training mixes: ~160K PDB structures with a May 2020 cutoff (as in ESMFold), ~270K high‑confidence SwissProt distilled structures from AFDB, and ~1.9M high‑confidence cluster representatives from AFESM, totaling roughly 2M structures.
The largest 3B model uses an extended AFESM‑E, sampling up to 10 proteins per cluster (pLDDT ≥ 80), reaching ~8.6M distilled structures, then combined with PDB and SwissProt. This scale lets a general‑purpose architecture absorb folding regularities without explicit pair/triangle modules.
Why This Breakthrough Matters
SimpleFold fundamentally challenges the prevailing wisdom that high-accuracy protein folding requires intricate, domain-specific architectures. The conventional approach, epitomized by AlphaFold2 and similar systems, relies heavily on MSA processing, pair representations, and triangle updates.
If comparable accuracy can be achieved through a simpler, transformer-only design that scales with data and compute, it opens exciting possibilities for more flexible, accessible folding systems.
This shift is particularly valuable for challenging scenarios where traditional methods struggle. Orphan proteins, which lack sufficient homologous sequences for effective MSA construction, represent one such case.
A generative formulation also aligns better with the underlying physics: proteins exist as dynamic ensembles of conformations around low-energy states, not as rigid, single structures.
From a practical standpoint, SimpleFold's streamlined architecture offers significant advantages. The model is easier to maintain, port across different hardware platforms, and deploy in resource-constrained environments.
The smallest variant runs efficiently on consumer-grade devices, democratizing access to protein structure prediction for basic research and screening applications. Moreover, because the model inherently generates conformational ensembles, it naturally suits problems requiring structural diversity such as cryptic binding site discovery, which pose challenges for deterministic regression approaches.
Results and Performance Analysis
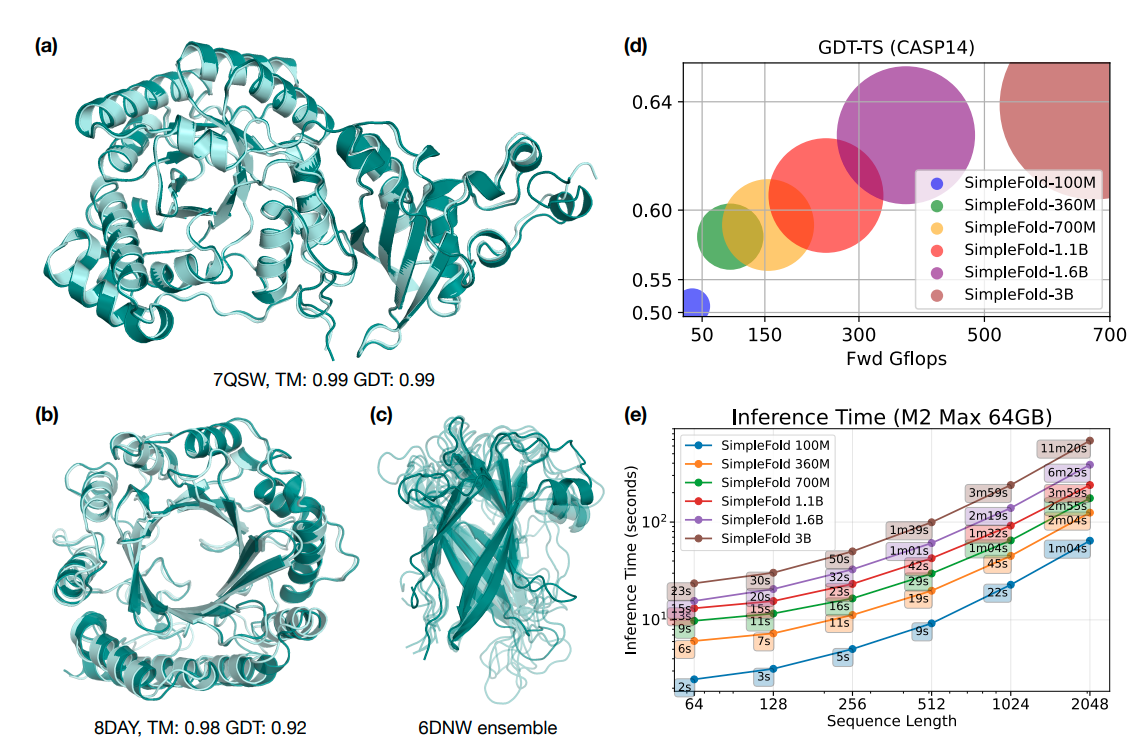
Figure 1 Example predictions of SimpleFold on targets (a) chain A of 7QSW (RubisCO large subunit) and (b) chain A of 8DAY (Dimethylallyltryptophan synthase 1), with ground truth shown in light aqua and prediction in deep teal. (c) Generated ensembles of target chain B of 6NDW (Flagellar hook protein FlgE) with SimpleFold finetuned on MD ensemble data. (d) Performance of SimpleFold on CASP14 with increasing model sizes from 100M to 3B. (e) Inference time of different sizes of SimpleFold on consumer level hardware, i.e., M2 Max 64GB Macbook Pro.
Figure 1 (above) provides an excellent visual overview of SimpleFold's capabilities: example predictions on diverse targets, ensembles after MD fine‑tuning, a size‑performance curve on CASP14 (100M -> 3B), and inference timings on a MacBook Pro M2 Max (64 GB). The central message is that a standard transformer architecture, using flow matching, can compete with highly specialized folding systems and benefits significantly from increased scale.
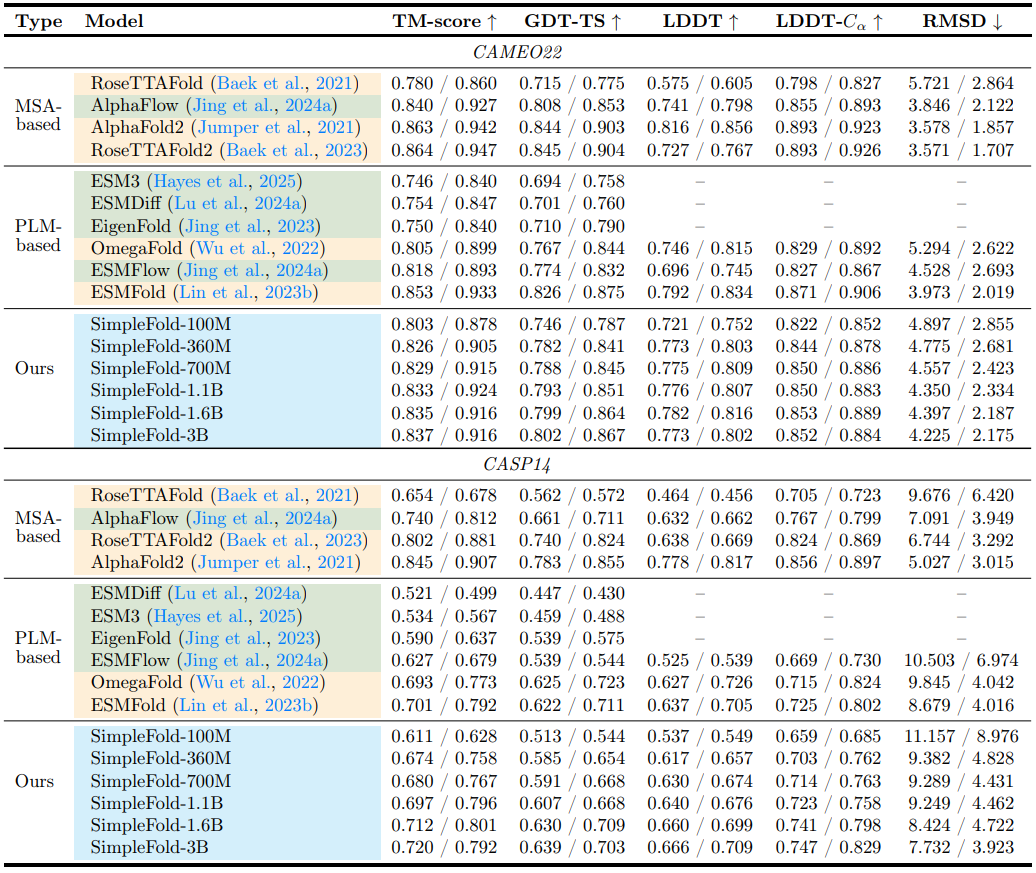
Table 1 Performance of protein folding on the CAMEO22 (top) and CASP14 (bottom) benchmarks. For each metric, we report the average / median over all samples. Here, orange denotes baselines trained with regression objectives, green denotes baselines trained with generative objectives (i.e., diffusion/flow-matching or autoregression), and blue denotes our SimpleFold, which is trained with generative objective but without MSA.
Table 1 (above) reports CAMEO22 and CASP14 results. On CAMEO22 (Haas et al., 2018), SimpleFold's overall performance is comparable to strong baselines (ESMFold, RoseTTAFold2, AlphaFold2). On CASP14 (Pereira et al., 2021), SimpleFold‑3B outperforms ESMFold on TM‑score (0.720/0.792 vs. 0.701/0.792) and GDT‑TS (0.639/0.703 vs. 0.622/0.711), showing that a PLM‑conditioned, MSA‑free, generative architecture can match or beat a state‑of‑the‑art regression baseline. The paper notes that "pure generative" models can sometimes lag on single‑target benchmarks that favor point‑estimates, yet SimpleFold narrows or flips that gap on CASP14.
Beyond single structures, SimpleFold's ensemble generation is a differentiator. Section 4.4 tests on the ATLAS MD ensemble dataset (1390 proteins; 250 generated conformations per test protein). Without additional tuning, SimpleFold produces ensembles that align better with MD distributions than MSA‑based baselines like AlphaFold2 with MSA‑subsampling.
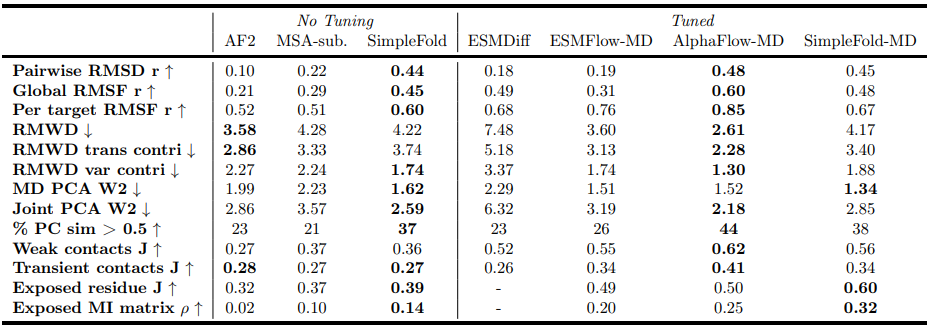
Table 2 Evaluation on MD ensembles. Results of baseline models are taken from (Jing et al., 2024a; Lu et al., 2024a), to which the evaluation pipeline for our SimpleFold (SF) and SimpleFold-MD (SF-MD) adheres
With a short fine‑tune (SimpleFold‑MD), the model improves further and compares favorably to contemporaries that also tune on MD (e.g., reported gains versus ESMFlow‑MD and AlphaFlow‑MD on ensemble observables). Table 2 provides the metric breakdown across flexibility (RMSD r, RMSF r), distributional distances (RMWD), PCA‑based Wasserstein distances, and contact/solvent exposure observables.
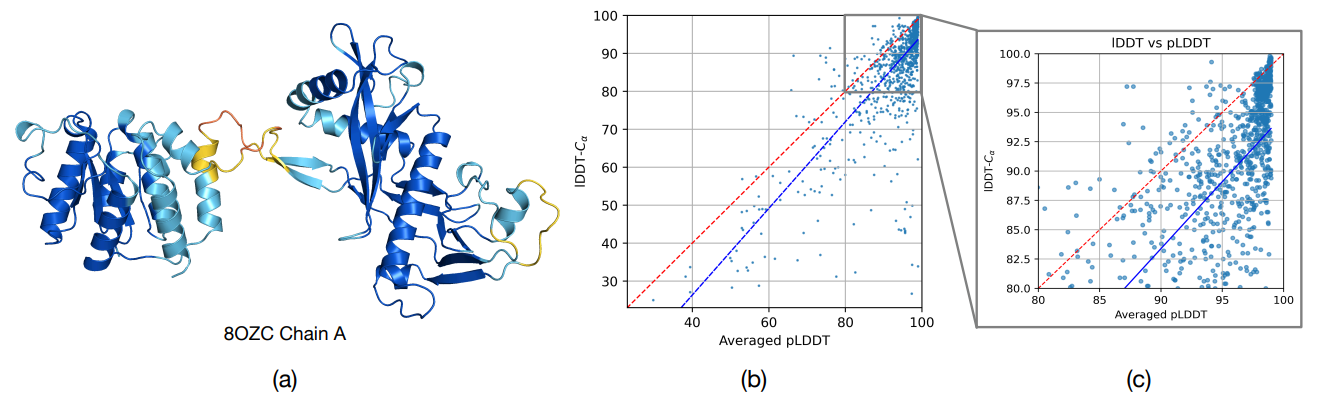
Figure 3 (a) An example prediction of SimpleFold with pLDDT (color red to dark blue denote pLDDT low to high following visualization from Chakravarty and Porter (2022)). (b) & (c) Comparison of pLDDT and LDDT-Cα.
Figure 3 (above) studies SimpleFold's confidence head. A lightweight pLDDT module (trained on discretized LDDT targets) achieves a Pearson correlation of 0.77 with LDDT‑Cα across CAMEO22 and additional PDB chains after 2023. That correspondence indicates the module's utility for quality estimation, and since it's decoupled from sampling, it can plausibly score structures produced by other models.
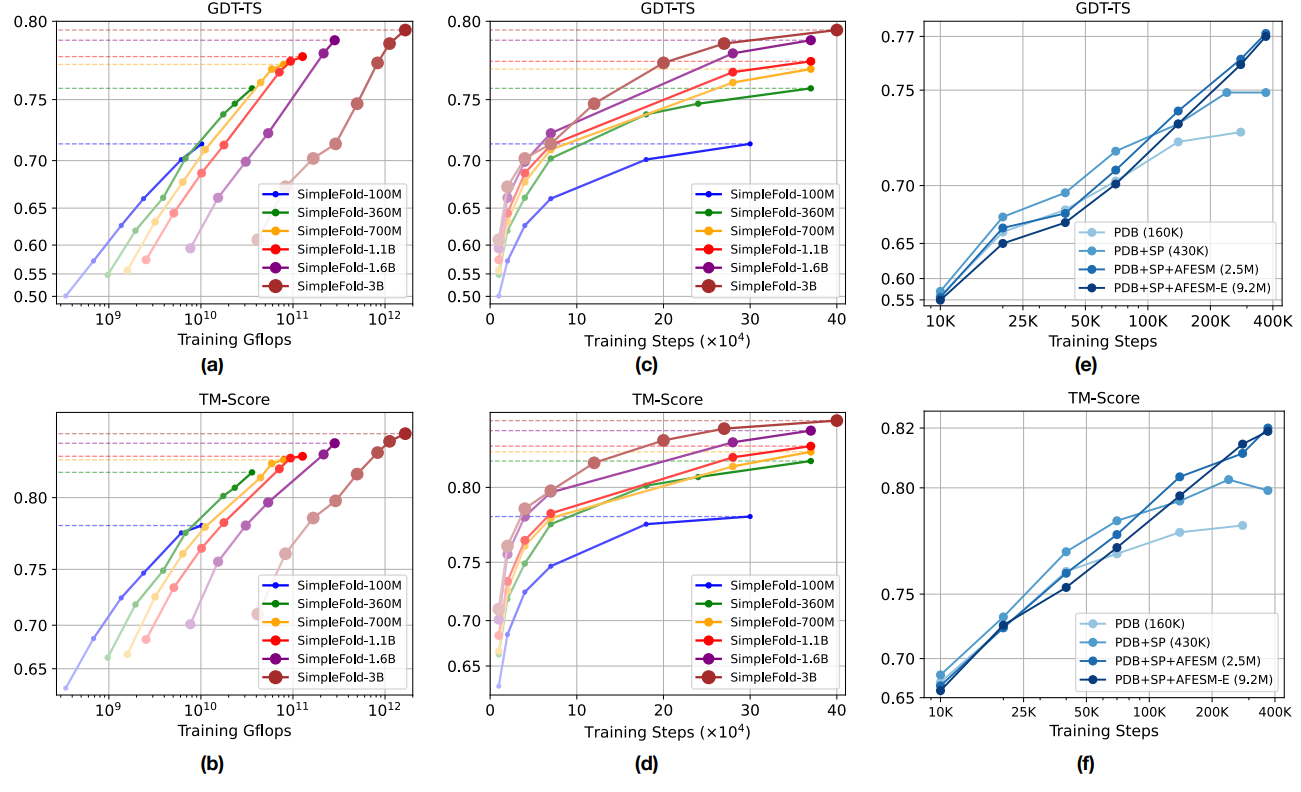
Figure 4 Scaling behavior of SimpleFold. Training Gflops vs. folding performance on GDT-TS and (b) TM-score. Training steps vs. folding performance on (c) GDT-TS and (d) TM-score. How data scale affects the performance (e) GDT-TS and (f) TM-score. All models are benchmarked on CAMEO22.
Figure 4 (above) analyzes scaling. Training compute (Gflops) increases with model size, while CAMEO22 performance on GDT‑TS and TM‑score rises accordingly. The improvements are especially pronounced on CASP14-consistent with the idea that higher‑capacity generative models better capture long‑range dependencies and tertiary organization needed by harder targets. The paper's ablations in the appendix further examine timestep resampling (biasing toward late‑time steps closer to data), LDDT loss weighting, and inference hyperparameters.

Figure 11 Examples of failure cases (TM-score < 0.6) of SimpleFold predictions with ground truth shown in light aqua and prediction in deep tea (first row from CAMEO22 targets and second row from CASP14 targets). We also include predictions from ESMFold for comparison.
Figure 11 (appendix) shows failure cases (TM‑score < 0.6), emphasizing that certain topologies remain challenging. The authors suggest that additional scale, refined training schedules, or hybridization with lightweight geometric priors could help.
Training Data and Methodology
SimpleFold's training strategy combines empirical structures with large-scale distilled predictions. The base models train on approximately 2M structures: ~160K PDB entries (May 2020 cutoff, matching ESMFold's training set), ~270K high-quality AFDB SwissProt entries (average pLDDT > 85, standard deviation < 15), and ~1.9M AFESM cluster representatives with pLDDT > 0.8.
The largest 3B parameter model leverages an extended AFESM-E dataset, sampling up to 10 proteins per cluster (average pLDDT ≥ 80) to reach ~8.6M distilled structures when combined with PDB and SwissProt data.
For sequence conditioning, the model uses frozen ESM2-3B per-residue embeddings concatenated with learned residue tokens. The training objective combines flow-matching loss with a local LDDT term, while inference integrates the learned velocity field with optional Langevin noise for stochastic sampling.
The evaluation framework relies on established protein folding benchmarks. CAMEO22 and CASP14 provide comprehensive folding assessments using standard metrics: TM-score and GDT-TS (measuring global topology), LDDT (local distance difference test) and LDDT-Cα (local accuracy), and RMSD (global distance; lower values indicate better performance).
For ensemble evaluation, the ATLAS dataset supplies MD-derived conformational distributions across numerous proteins, enabling assessment of flexibility correlations, Wasserstein distances in PCA subspaces, and contact/solvent exposure statistics.
Limitations and Trade-offs
SimpleFold's general-purpose design philosophy involves important trade-offs. By eschewing explicit geometric biases in favor of data and compute scaling, the model places greater demands on training data quality and optimization schedules.
Techniques like timestep resampling (biased toward later time steps to improve side-chain prediction) become more critical for achieving optimal performance. While the results are competitive overall, some individual targets still underperform against specialized systems, and CASP14 remains challenging across all approaches.
Additionally, ensemble generation quality shows sensitivity to sampling hyperparameters particularly the noise scale
Conclusion
SimpleFold represents an important experiment in protein folding methodology, demonstrating that high-quality structural prediction doesn't require the complex, domain-specific architectures that have dominated the field.
By achieving competitive single-structure accuracy on CAMEO22 and CASP14 benchmarks while excelling at ensemble generation, this transformer-only, flow-matching approach opens new possibilities for the field.
The 100M parameter variant offers an attractive solution for resource-constrained environments, while the 3B parameter model shows clear advantages on challenging targets where increased model capacity pays dividends.
Looking ahead, this work establishes a foundation for numerous exciting research directions. Future investigations could explore even larger model scales, incorporate lightweight geometric constraints to boost performance on difficult targets, or adapt the framework for downstream applications like protein design and drug docking.
Perhaps most importantly, SimpleFold's simplified architecture and strong ensemble generation capabilities position it well for the growing emphasis on conformational diversity in structural biology and pharmaceutical research.
Definitions
TM‑score: Scale‑invariant measure of global structural similarity between two protein structures (0–1; higher is better).
GDT‑TS: Global Distance Test—Total Score; percentage of residues within preset distance thresholds after superposition (0–1; higher is better).
LDDT: Local Distance Difference Test; per‑residue/local accuracy measure (0–100 or 0–1; higher is better).
MSA: Multiple sequence alignment; set of homologous sequences used to extract evolutionary constraints.
Flow matching: Training a time‑dependent velocity field to transform noise into data along a pre‑defined path, enabling efficient generative sampling.
ESM2‑3B: A 3B‑parameter protein language model used here as a frozen sequence encoder
References
AlphaFold2: (Jumper et al., 2021).
RoseTTAFold: (Baek et al., 2021).
ESM2: (Lin et al., 2023).
Flow Matching: (Lipman et al., 2022);
Stochastic Interpolants: (Albergo and Vanden‑Eijnden, 2023).
CAMEO: (Haas et al., 2018).
CASP14 overview: (Pereira et al., 2021). SimpleFold paper: (Wang et al., 2025).


SimpleFold: When Less Is More in Protein Folding
SimpleFold: Folding Proteins is Simpler than You Think