The brevianamide alkaloids represent a structurally fascinating subset of fungal diketopiperazine dimers, with most family members featuring characteristic tryptophan-tryptophan oxidative linkages.
Brevianamide S stands as a notable outlier, incorporating an unusual proline-proline connection between two unsaturated diketopiperazine cores. Isolated from marine Aspergillus versicolor MF030, this natural product demonstrated selective antitubercular activity against M. bovis BCG (MIC 6.25 μg/mL) without significant broad-spectrum antibacterial effects, suggesting a specialized mode of action worthy of synthetic investigation ((Song et al., 2012)).
Lawrence and coworkers now report the total synthesis of Brevianamide S through an ambitious bidirectional strategy that showcases two key methodological advances: a copper-promoted alkenyl-alkenyl Stille coupling between dehydroproline-derived fragments, and a bis-lactim ether activation protocol enabling efficient double aldol condensations ((Lockyer et al., 2025)).
This convergent approach not only provides synthetic access to an architecturally unique natural product but establishes a modular platform for systematic structure-activity relationship (SAR) studies within this underexplored dimer class.
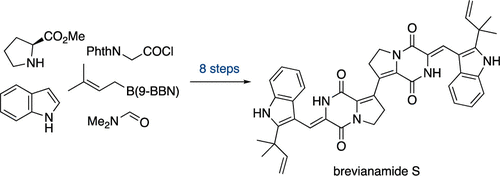
Image Credit Lockyer et al.
Key Takeaways
- First total synthesis of brevianamide S achieved in an eight-step longest linear sequence via a bidirectional plan.
- Preparation of a key alkenyl iodide on a diketopiperazine ring in three chromatography-free steps, enabling scalable access to coupling partners.
- Bespoke alkenyl-alkenyl Stille coupling, accelerated by a copper(I) additive, for C=C bond formation between two unsaturated diketopiperazines, consistent with known copper effects in Stille chemistry ((Farina et al., 1994)).
- Double aldol condensation proceeds efficiently only after converting both lactams to methyl lactim ethers to enhance solubility and acidity at the reacting methylene positions ((Schoellkopf, 1983); (Gonzalez et al., 2004)).
- Final deprotection of methyl lactim ethers to lactams completes the target, with spectroscopic data matching the original natural product.
- Access to this rare proline-proline dimer architecture offers a tractable scaffold for antitubercular lead exploration and SAR, complementing prior total syntheses across the brevianamide family ((Godfrey et al., 2020); (Godfrey et al., 2022)).
Cross-Coupling Chemistry And Mechanistic Insights
The central C-C bond formation between alkenyl iodide fragments represents a particularly challenging transformation given the electron-deficient nature and limited solubility of diketopiperazine substrates. Initial attempts with classical Ullmann homocoupling and Heck-type protocols proved unproductive, likely due to competing coordination of the heterocyclic nitrogens and poor substrate solubility under standard conditions.
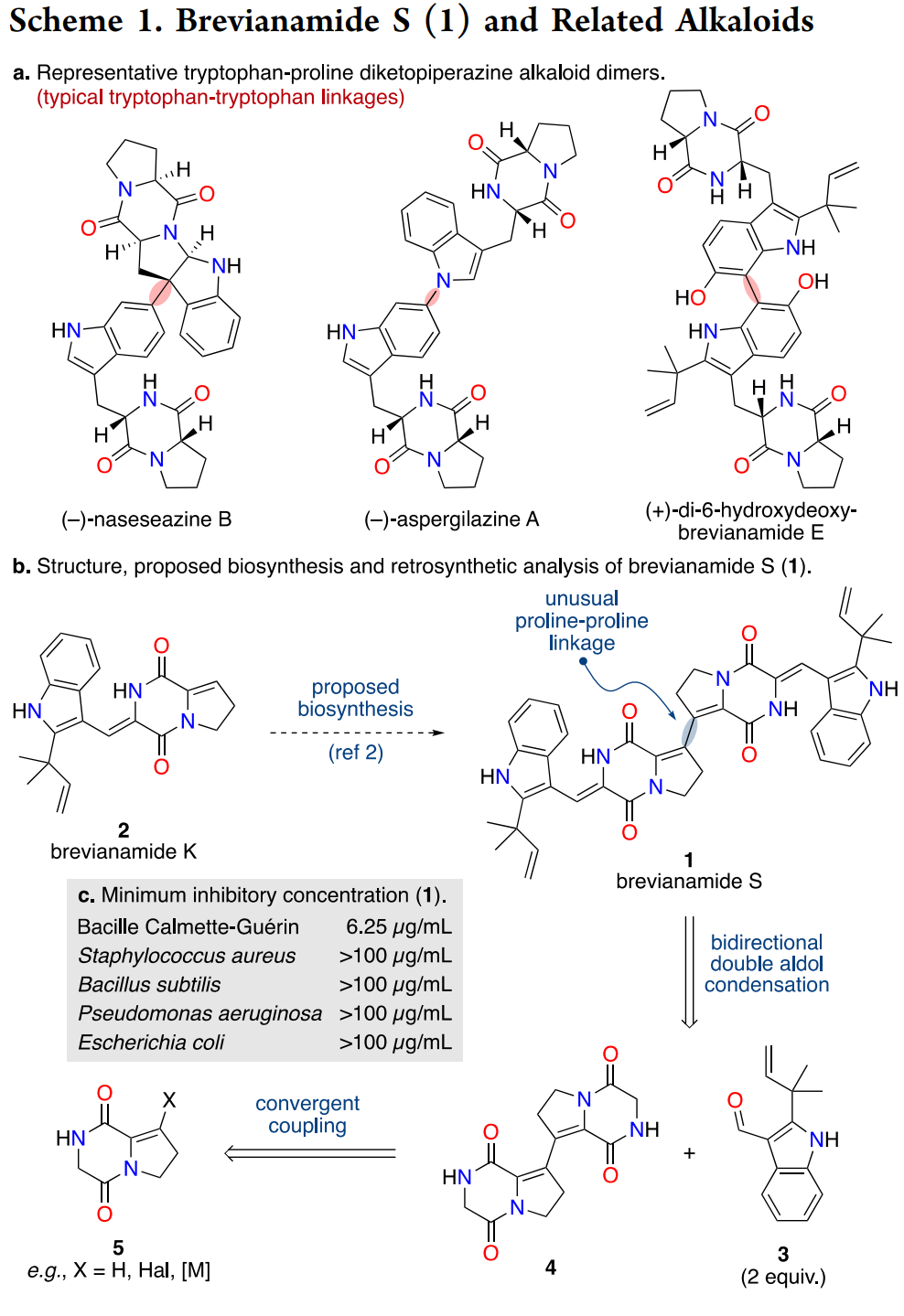
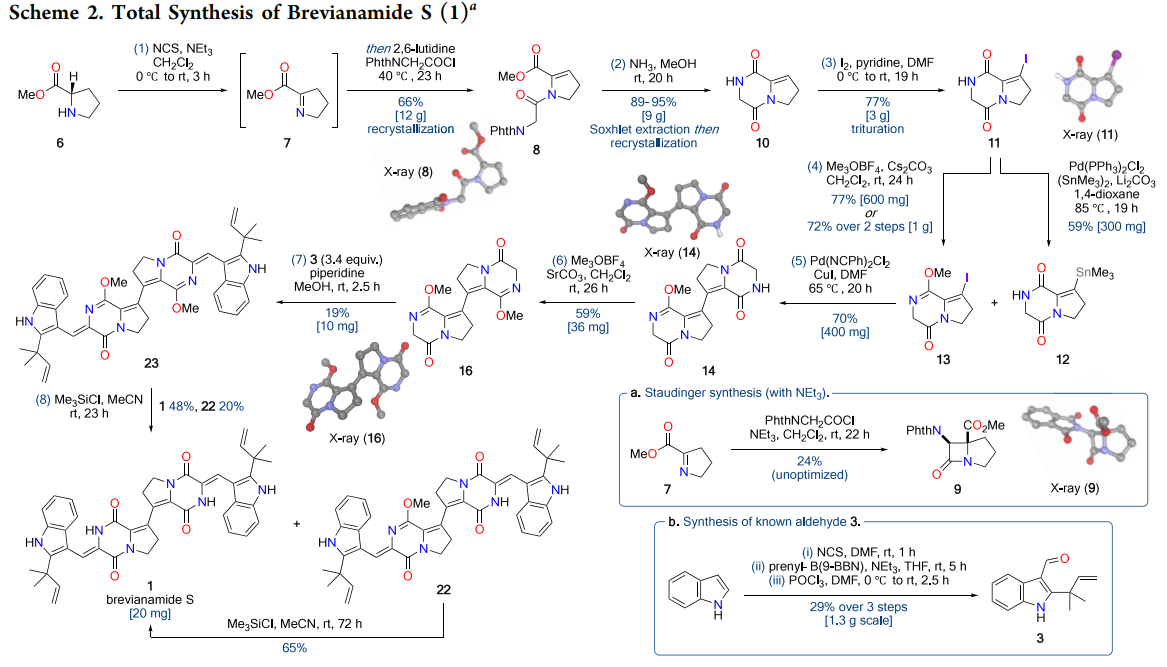
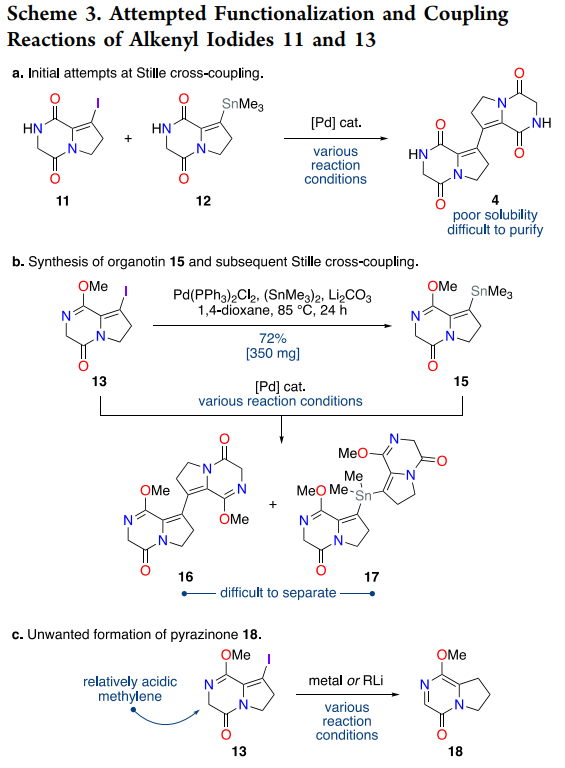
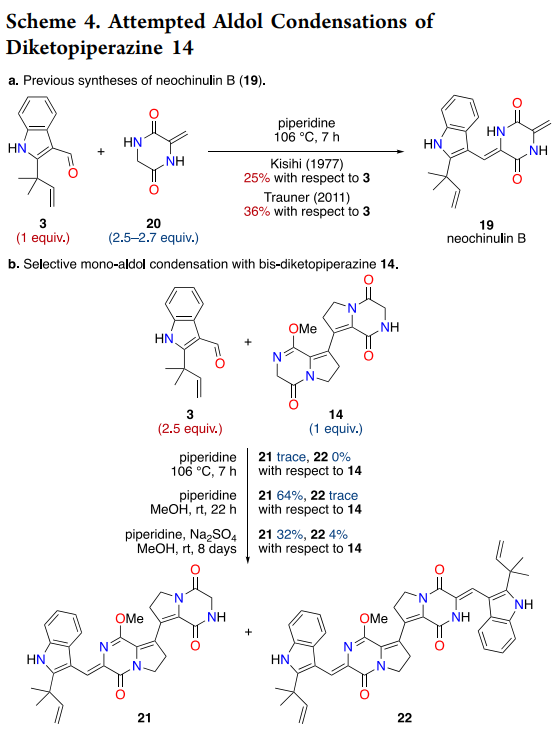
Image Credits Lockyer et al.
The breakthrough came through Stille methodology, leveraging the pre-formed organostannane 12 (accessible in 59% yield via palladium-catalyzed stannylation of iodide 11). However, direct coupling between iodide 11 and stannane 12 suffered from the extreme insolubility of the resulting bis-diketopiperazine product 4. This challenge was elegantly circumvented by converting one coupling partner to methyl lactim ether 13, which dramatically improved solubility while maintaining coupling competency.
The optimized Stille protocol employs bis(benzonitrile)palladium dichloride as precatalyst with copper(I) iodide as additive, delivering bis-diketopiperazine 14 in 70% yield on >400 mg scale. The copper effect in Stille chemistry is well-established: Cu(I) species facilitate transmetalation from tin to palladium through organocopper intermediates, effectively accelerating the rate-limiting step while suppressing competing β-hydride elimination pathways ((Farina et al., 1994)). This additive proves particularly beneficial for challenging heteroaryl and alkenyl substrates where electronic factors retard standard transmetalation.
Aldol Strategy And Lactim Ether Activation
The installation of both pendant side chains through double aldol condensation represents perhaps the most ambitious aspect of the synthesis. Historical precedent from the Kishi and Trauner groups on related systems suggested that DKP aldol reactions typically suffer from modest yields (25-36%) even with excess nucleophile, making a symmetric double aldol particularly challenging (Kuttruff et al., 2011).
The key insight emerged from lactim ether chemistry pioneered by Schöllkopf for amino acid synthesis ((Schoellkopf, 1983)). Conversion of the secondary lactams to methyl imidates serves multiple functions: enhanced solubility, increased α-methylene acidity through imidate stabilization of the resulting enolate, and reduced catalyst coordination issues. Initial studies on mono-lactim ether 14 with aldehyde 3 demonstrated selective condensation adjacent to the activated site, yielding monoadduct 21 in 64% yield while double condensation remained inefficient (4% yield).
Formation of bis-lactim ether 16 proved decisive, enabling double aldol condensation under mild conditions (piperidine, MeOH, ambient temperature) to deliver dimethyl-brevianamide S (23) in 19% overall yield—equivalent to ~44% per aldol event, representing a significant improvement over literature precedent. Final lactim ether deprotection using trimethylsilyl chloride proceeded through standard silicon-mediated O-alkyl cleavage to regenerate the natural lactam functionality ((Jung and Lyster, 1977)).
Synthetic Applications And Medicinal Chemistry Potential
Beyond providing the first synthetic access to brevianamide S, this route establishes a modular platform uniquely suited for systematic structure-activity relationship studies.
The brevianamide alkaloids have emerged as privileged scaffolds for complex synthesis methodology development, with recent landmark contributions including the first total synthesis of brevianamide A and unified biosynthetic approaches to multiple family members ((Godfrey et al., 2020); (Godfrey et al., 2022)).
The selective antitubercular profile of brevianamide S against BCG, coupled with minimal broad-spectrum activity, suggests a specialized mechanism potentially exploiting mycobacterial-specific vulnerabilities ((Song et al., 2012)).
Given the urgent need for novel antitubercular agents with distinct modes of action to combat emerging resistance ((Dartois and Rubin, 2022)), the proline-proline dimer architecture represents an underexplored chemotype with significant therapeutic potential.
The convergent nature of the synthetic strategy allows for systematic variation at multiple positions: electronic modulation through side chain substitution, geometric constraints via aldol stereochemistry, and core modifications through alternative diketopiperazine precursors.
The scalable preparation of key intermediates (multigram quantities achievable without chromatography) and the room-temperature aldol protocol make this approach particularly amenable to parallel synthesis campaigns.
Why It Is Important
The brevianamide family has been a proving ground for complex alkaloid synthesis and biosynthetic hypothesis testing, including a recent first total synthesis of brevianamide A and a unified access route to family members ((Godfrey et al., 2020); (Godfrey et al., 2022)).
Brevianamide S adds a rare proline-proline dimer topology to that canon. The current strategy accomplishes two things at once: it provides authentic material for biological testing and it sets up a practical platform for analogue libraries that tune electronics, geometry, and substitution around the central C3dC and the side chains.
From a biomedical perspective, the original isolation paper reported selective inhibition of BCG, a standard surrogate for M. tuberculosis screens ((Song et al., 2012)). The TB drug discovery field continues to need new chemotypes with novel modes of action to address resistance and long treatment courses ((Dartois and Rubin, 2022)).
Synthetic control over a proline-proline linked diketopiperazine dimer gives medicinal chemists a foothold to probe SAR and potentially identify tractable targets, even if the present work is purely synthetic.
In parallel, the chemical technology is broadly useful: scalable iodination on a DKP, reliable alkenyl-alkenyl dimerization under Stille conditions, and a generalizable double aldol tactic on bis-lactim ethers.
Discussion
Scheme analysis and reaction optimization.
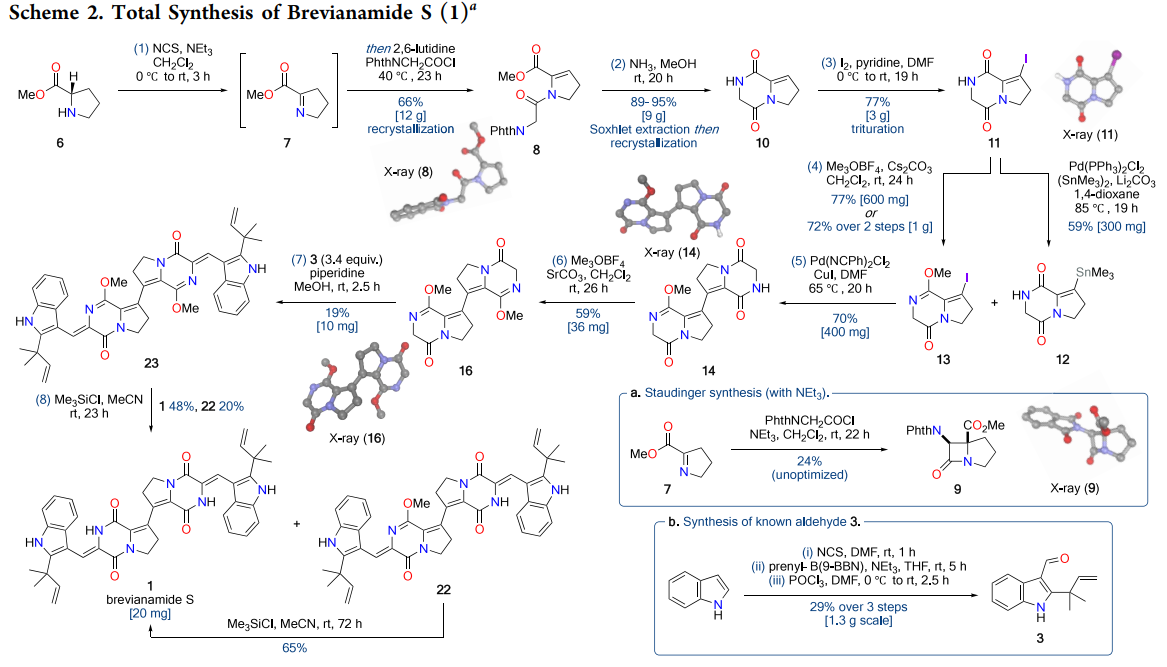
The synthetic schemes reveal several key strategic decisions that proved crucial for success. Scheme 2 outlines the complete eight-step sequence, highlighting the chromatography-free preparation of alkenyl iodide 11 and the scalable Stille dimerization. Particularly noteworthy is the base-dependent selectivity in the initial enamide formation—stronger bases like triethylamine promote a competing [2+2] Staudinger cyclization, necessitating the use of 2,6-lutidine for clean N-acylation.
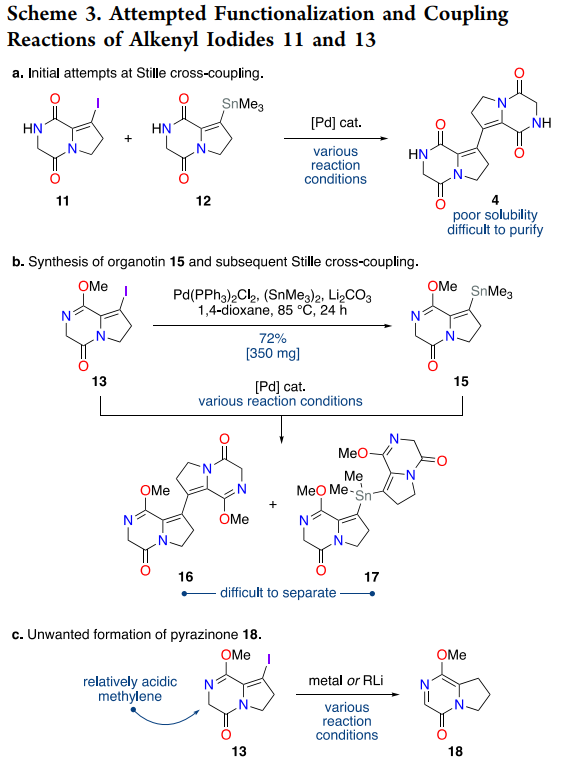
Scheme 3 documents the extensive optimization required for the cross-coupling step, with numerous failed metalation attempts leading to pyrazinone formation via intramolecular imidate condensation. This side reaction provided valuable mechanistic insight into lactim ether reactivity under strongly basic conditions and ultimately guided the selection of Stille methodology over alternative cross-coupling approaches.
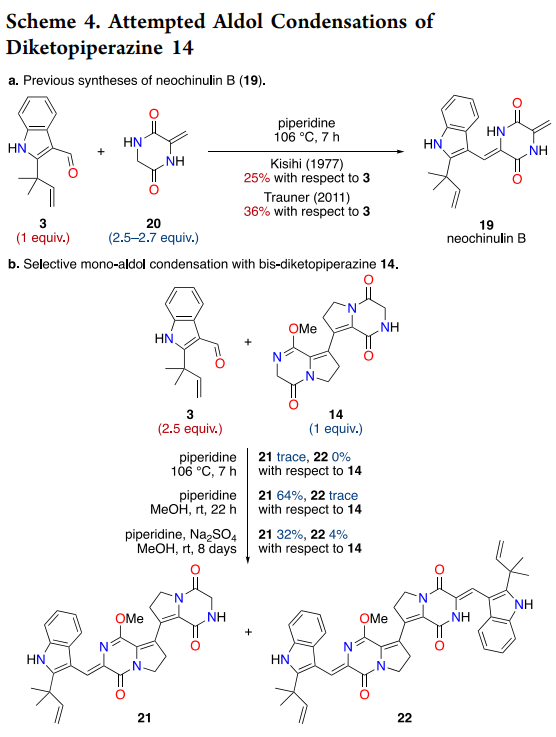
Scheme 4 elegantly demonstrates the power of lactim ether activation in the aldol sequence. The dramatic difference in double aldol efficiency between bis-lactam 14 (essentially unreactive) and bis-lactim ether 16 (19% yield) underscores the importance of substrate pre-activation for challenging transformations on sterically encumbered heterocycles.
Step economics and scalability.
The alkenyl iodide intermediate (11) is prepared in three steps without chromatography on multigram scale, a nontrivial practical win in complex heterocycle chemistry. While Ullmann and Heck variants struggled, the chosen Stille protocol with bis(benzonitrile)palladium dichloride and copper(I) iodide delivered hundreds of milligrams of the bis-DKP in a single batch, highlighting a reliable C=C forging step. The double aldol is formally only 19 percent overall yield to the dimethyl-protected product, but that corresponds to approximately 44 percent yield per aldol event, in line with and in some cases better than literature precedents for DKP aldol chemistry (Gonzalez et al., 2004).
Reaction mechanism insights.
The iodination potentially proceeds via enolization of the unsaturated DKP followed by electrophilic iodination of the activated alkene or vinylic position under I32 in pyridine, a standard route to alkenyl iodides on electron-deficient systems. For the Stille coupling, the copper effect is well documented: copper(I) salts can form organocopper intermediates that transmetalate more readily to palladium, accelerating the coupling and improving yields for challenging substrates like heteroaryl and alkenyl partners ((Farina et al., 1994)). Given the low solubility of bis-DKPs, the authors also improved coupling efficiency by first converting one partner to a methyl lactim ether, which increases solubility and moderates coordination to the metal center, reducing catalyst deactivation.
For the aldol sequence, methyl lactim ethers enhance the acidity at the alpha position by replacing an amide carbonyl with its imidate analogue, stabilizing the resulting enolate and directing addition. Under methanolic, piperidine-mediated conditions at room temperature, a single aldol occurred selectively adjacent to the lactim ether on a mixed DKP. Converting both lactams to lactim ethers made the double aldol feasible, possibly by providing two similarly activated sites that could each enolize and add to the aldehyde. This logic aligns with classic bis-lactim ether reactivity patterns originated by Schoellkopf for enantioselective alpha-functionalization ((Schoellkopf, 1983)) and with improvements in DKP aldol chemistry using carefully chosen bases and solvents ((Gonzalez et al., 2004)).
The observed formation of a pyrazinone byproduct upon attempts at metalation or lithium-halogen exchange on an iodinated lactim ether likely arises from intramolecular condensation of an activated alpha position with the neighboring imidate, followed by rearrangement to a more stable aromatic-like pyrazinone core. While the present paper does not detail the mechanism, the outcome is consistent with the increased electrophilicity and lability of methyl lactim ethers under strong bases, and it explains why the authors favored cross-coupling routes that avoided such conditions.
Finally, deprotection of methyl lactim ethers back to secondary lactams was achieved with trimethylsilyl chloride in acetonitrile. Silicon-based dealkylations of ethers are well established, with trimethylsilyl reagents or in situ generated silyl halides cleaving alkyl-oxygen bonds under mild conditions to regenerate carbonyl derivatives ((Jung and Lyster, 1977); (Prendergast and McGlacken, 2017)). The reported yields and the clean match of spectral data to the natural product confirm the fidelity of the final step.
Comparisons and context in the brevianamide family.
Prior syntheses in the family have showcased pericyclic logic and biomimetic steps, for example in the variecolortide series and in total syntheses that tested biosynthetic sequences ((Kuttruff et al., 2011); (Kuttruff et al., 2012)). The present route is less about biosynthetic emulation and more about platform building: it standardizes three moves that can be mixed-and-matched for analogues, namely DKP iodination, alkenyl-alkenyl Stille dimerization, and bis-lactim-ether double aldol.
Conclusion
Lawrence and coworkers have achieved an elegant inaugural synthesis of brevianamide S that advances both target-oriented synthesis and methodology development. The eight-step route showcases three enabling technologies: chromatography-free preparation of functionalized diketopiperazines, copper-promoted Stille coupling of challenging alkenyl partners, and lactim ether-activated double aldol condensations under mild conditions. Each methodological advance addresses fundamental challenges in heterocycle functionalization while maintaining practical scalability.
The strategic significance extends beyond the immediate target to encompass broader applications in alkaloid synthesis and medicinal chemistry. The modular design enables systematic structural variation around the proline-proline dimer core, positioning this approach as a valuable platform for antitubercular lead optimization. With authentic material now accessible for biological evaluation and a proven synthetic pathway for analogue generation, the stage is set for comprehensive structure-activity relationship studies within this architecturally unique natural product class.
Synthetic chemists will find particular value in the mechanistic insights surrounding lactim ether reactivity, the copper-enhanced Stille protocol for challenging substrates, and the strategic use of protective group manipulation to enable otherwise intractable transformations. This work exemplifies how thoughtful synthetic planning can overcome inherent substrate limitations while establishing broadly applicable methodological advances.
Definitions
Stille coupling: A palladium-catalyzed cross-coupling between an organostannane and an organic electrophile; copper(I) salts can accelerate transmetalation via formation of organocopper intermediates ((Farina et al., 1994)).
Lactim ether activation: Conversion of lactams to methyl imidates that enhances α-methylene acidity and improves solubility; key enabling technology for difficult aldol condensations on diketopiperazines ((Schoellkopf, 1983)).
Aldol condensation on DKPs: Carbon-carbon bond formation via enolization adjacent to a DKP followed by addition to an aldehyde; traditionally low-yielding unless substrate activation and optimized conditions are employed ((Gonzalez et al., 2004)).
BCG surrogate screening: Use of attenuated M. bovis bacillus Calmette-Guérin as a biosafety-compatible surrogate for M. tuberculosis in early-stage antibacterial discovery ((Ahmed et al., 2021)).
Diketopiperazine (DKP): A cyclic dipeptide featuring a six-membered 2,5-diketopiperazine ring; a common scaffold in fungal alkaloids and a versatile synthon in organic chemistry.
Methyl lactim ether: The imidate analogue of a lactam formed by alkylation at the carbonyl oxygen; increases alpha acidity and alters solubility and coordination properties ((Schoellkopf, 1983)).
References: (Lockyer et al., 2025); (Song et al., 2012); (Godfrey et al., 2020); (Godfrey et al., 2022); (Farina et al., 1994); (Schoellkopf, 1983); (Gonzalez et al., 2004); (Huy et al., 2011); (Inoue et al., 1977); (Dartois and Rubin, 2022); (Ahmed et al., 2021); (Kuttruff et al., 2011); (Kuttruff et al., 2012); (Prendergast and McGlacken, 2017)


Building a Molecule to Fight Tuberculosis: The Total Synthesis of Brevianamide S
Total Synthesis Of Brevianamide S