A groundbreaking study published in Nature Communications by researchers from Zhejiang Lab, Shanghai Jiao Tong University, and ShanghaiTech University has leveraged large language models to systematically discover novel CRISPR-Cas proteins, specifically identifying seven previously unknown subtypes of Cas12a proteins with unique structural and functional characteristics.
The CRISPR-Cas systems have revolutionized molecular biology and biotechnology since their discovery as bacterial adaptive immune systems (Jinek et al., 2012). These systems offer unprecedented precision in gene editing and detection applications, building on foundational work in nucleic acid sequence analysis (Altschul et al., 1990).
The research team, led by Peixiang Ma, Xingxu Huang, Jin Tang, and Xiaokang Zhang, developed an Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan) strategy based on Meta's Evolutionary Scale Model (ESM), a transformer-based language model trained on protein sequences.
This approach represents an important advancement from traditional sequence similarity-based methods to AI-driven protein discovery, enabling the identification of functionally distinct Cas proteins that might be missed by conventional approaches.
Key Takeaways
- Development of AIL-Scan, an AI system achieving 98.22% accuracy in CRISPR-Cas protein identification using ESM-2 15B parameter model
- Discovery of seven new Cas12a subtypes with unique CRISPR loci organizations and distinct integrase protein (an enzyme that facilitates the integration of viral DNA into the DNA of a host cell ) combinations
- Identification of eight structural variants each for Cas1, Cas2, and Cas4 integrase proteins through AlphaFold2 modeling
- Characterization of novel PAM recognition patterns, including AmCas12a's broader G-start PAM preference (PAM: a short DNA sequence (typically 2-6 base pairs) that is located immediately downstream of the target DNA)
- Demonstration of sensitive oncogene Single Nucleotide Polymorphism (SNP) detection capabilities without traditional TTTV PAM requirements
- Cryo-EM structural analysis revealing unique RNA-protein interactions in AmCas12a at 2.9 Å resolution
Revolutionary Approach to Protein Discovery
Traditional methods for discovering CRISPR-Cas proteins rely heavily on sequence similarity searches using tools like BLAST (Altschul et al., 1990) and Hidden Markov Models (HMMs). These approaches are fundamentally limited by their dependence on known sequences and struggle to identify truly novel proteins with divergent sequences but similar functions.
The researchers recognized that protein function is ultimately determined by three-dimensional structure rather than amino acid sequence alone, motivating their exploration of evolutionary scale language models.
The ESM-2 model, developed by Meta's FAIR team, represents a breakthrough in protein language modeling (Lin et al., 2023). Trained on 65 million protein sequences and scaling up to 15 billion parameters, ESM-2 can capture atomic-resolution features that traditional sequence-based methods miss.
The model learns the "language" of proteins, understanding how amino acid sequences fold into functional structures and recognizing patterns that indicate specific protein families and functions.
The AIL-Scan system fine-tuned ESM-2 on a carefully curated dataset of 76,567 non-redundant Cas proteins and 13,047 non-Cas proteins extracted from NCBI databases.
Using focal loss to address class imbalance, the system achieved remarkable performance: 97.75% accuracy with the 650M parameter model and 98.22% accuracy with the 15B parameter model.
Critically, the model maintained 97.68% accuracy on an independent test set containing sequences deposited after the training period, demonstrating robust generalization capabilities.
Why This Discovery Matters
The implications of this research extend far beyond academic interest in protein evolution. CRISPR-Cas12a proteins have emerged as powerful tools for gene editing, nucleic acid detection, and therapeutic applications due to their unique trans-cleavage activity.
Unlike Cas9, which requires both crRNA and tracrRNA, Cas12a systems use only crRNA and exhibit distinct cleavage patterns that make them valuable for specific applications.
The discovery of eight Cas12a subtypes with varying integrase compositions challenges our understanding of CRISPR system evolution and adaptation. Traditional CRISPR-Cas12a loci contain three integrase proteins: Cas1, Cas2, and Cas4, which are essential for incorporating new spacer sequences into CRISPR arrays.
However, the newly discovered subtypes show remarkable diversity, with some lacking all integrase proteins (subtype VIII) while others maintain selective combinations.
This structural diversity has profound functional implications. The researchers demonstrated that different subtypes exhibit distinct PAM (Protospacer Adjacent Motif) recognition patterns, metal ion preferences, and cleavage activities.
AmCas12a, for example, recognizes broader PAM sequences including G-start motifs, expanding the targeting range compared to conventional Cas12a proteins that prefer T-rich PAMs.
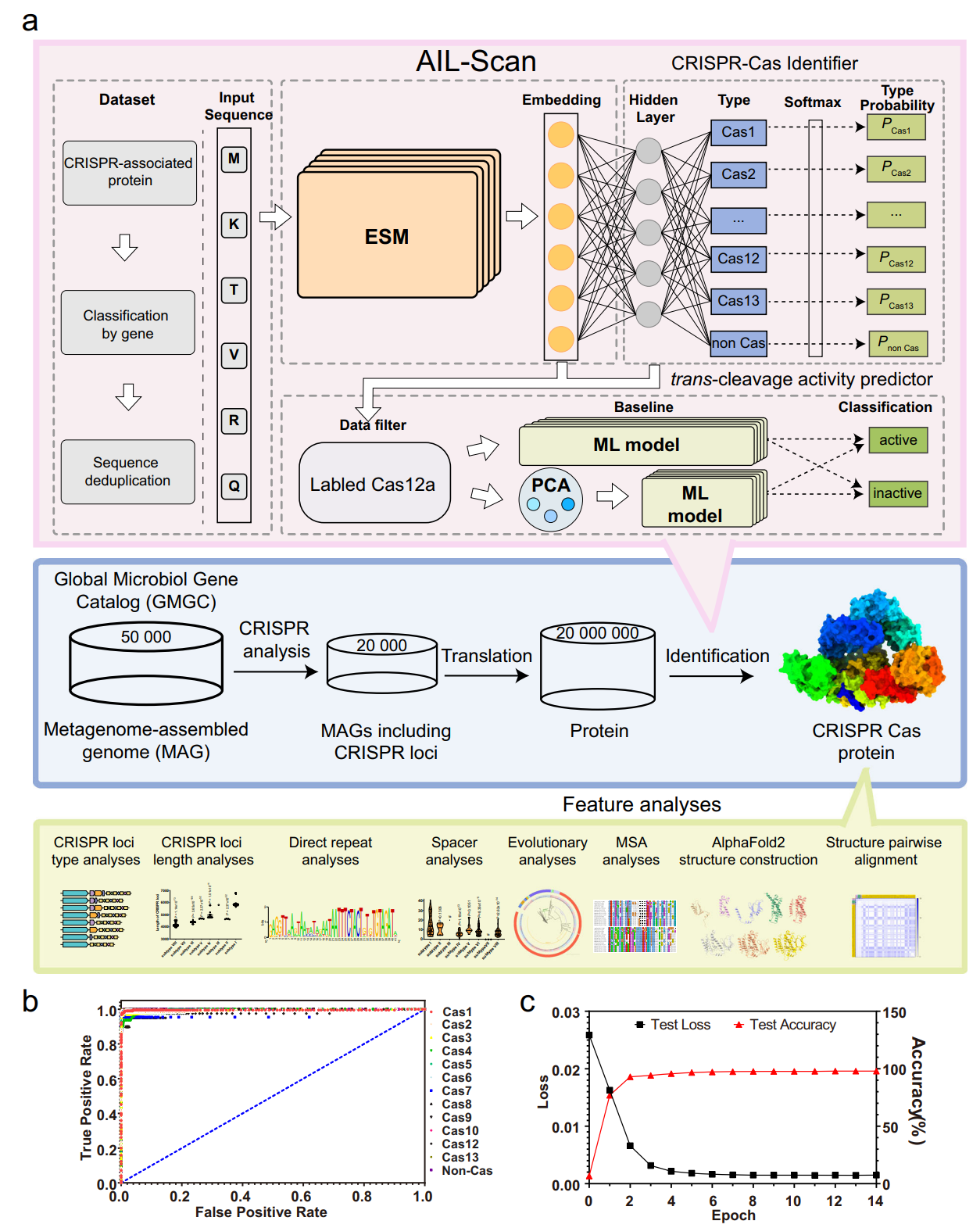
Fig. 1 | Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan). a The ESM language model is trained by Cas proteins, which were collected, classified, and clustered as input sequences. The Cas proteins were embedded and classified with multiple labels. The trans-cleavage activity prediction model was developed based on the ESM and small-scale experimental data of trans-cleavage. The trained model was applied to discover Cas proteins and predict features from the sequences extracted from the metagenome. The protein structures were visualized using Chimera59. The sequence alignment was visualized by Jalview61. b The receiver operating characteristic (ROC) curves and area under the ROC curve (AUC) for 12 Cas proteins and non-Cas proteins. c The test loss and test accuracy curves of AIL-Scan. Credit: Nature
Comprehensive Structural and Functional Analysis
The research team conducted extensive structural analyses using AlphaFold2 to predict protein structures for all discovered variants. The integrase proteins Cas1, Cas2, and Cas4 each showed eight distinct structural types, reflecting the evolutionary pressure to maintain function while adapting to different cellular environments.
Type 8 variants were most prevalent and resembled known structures, while other types showed unique features such as missing β-sheets or helices that could affect DNA binding and integration activities.
The Cas12a proteins themselves displayed remarkable structural diversity despite maintaining conserved catalytic domains. The RuvC and Nuc domains, responsible for DNA cleavage, retained critical residues across all subtypes, but surrounding regions showed significant variation. Root mean square fluctuation (RMSF) analysis revealed that while overall architecture remains conserved, specific regions exhibit subtype-specific adaptations.
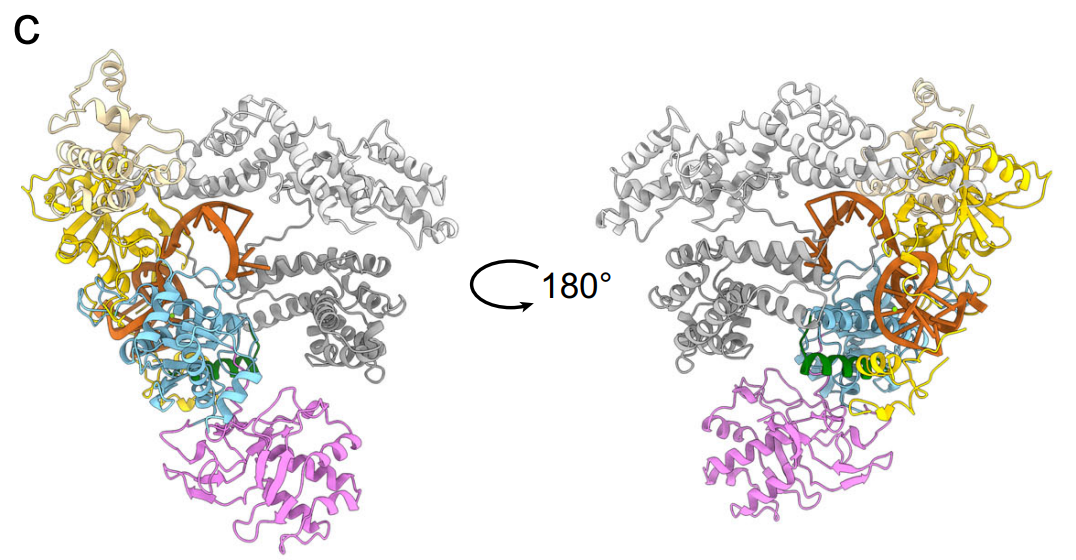
Fig. 6c | Structure of AmCas12a protein. The structure of AmCas12 revealed by cryoEM. (PDB: 8KGF, EMDB: EMD-37219) The structure alignments comparison with known Cas12a and other variants was analyzed in Supplementary Fig. 17. The structural domains were distinguished according to the color codes at the bottom Credit: Nature
Perhaps most striking was the cryo-EM structure of AmCas12a bound to crRNA at 2.9 Å resolution. This structure revealed an unprecedented RNA conformation where the spacer region forms an additional stem structure, stabilized by interactions with the WED, REC1, and RuvC domains. This unique configuration differs significantly from known Cas12a structures and suggests novel mechanisms for RNA-guided DNA targeting.
Functional characterization revealed that the newly discovered Cas12a variants exhibit diverse biochemical properties. While all variants demonstrated RNA and DNA binding capabilities, they showed differential preferences for divalent metal ions. Several variants, including AmCas12a and EvCas12a_2, preferred manganese over magnesium for optimal activity, potentially reflecting adaptation to specific cellular environments or targeting requirements.
Breakthrough Applications in Disease Detection
The practical applications of these discoveries became apparent when researchers tested the variants' abilities to detect clinically relevant genetic variations. Traditional CRISPR-Cas12a systems are limited by PAM requirements, restricting their utility for detecting certain disease-associated mutations.
The KRAS G12C oncogene mutation, a critical target for cancer diagnosis and treatment, lacks the conventional TTTV PAM sequence in its vicinity, making it inaccessible to standard Cas12a detection systems.
AmCas12a's broader PAM recognition proved instrumental in overcoming this limitation. The researchers designed crRNAs targeting the KRAS G12C mutation site and demonstrated that AmCas12a could specifically distinguish mutant sequences from wild-type with remarkable sensitivity.
The system detected as few as ten copies of the mutant sequence and could identify 0.1% mutant DNA in a wild-type background, surpassing the sensitivity of Sanger sequencing.
This capability represents a significant advancement in molecular diagnostics. Current detection methods for oncogene mutations often require complex sample preparation and expensive equipment. The AmCas12a system offers a potentially simpler, more cost-effective approach that could be adapted for point-of-care testing or resource-limited settings.
Technical Innovation and Model Interpretability
Beyond protein discovery, this research advances our understanding of how large language models process biological information. The researchers explored model interpretability by analyzing attention mechanisms within ESM-2, revealing that the model consistently focuses on catalytic domains critical for protein function.
Attention scores in cleavage domains were 2- to 24-fold higher than in non-cleavage regions, demonstrating that the model has learned to identify functionally important protein regions.
This interpretability analysis validates the biological relevance of the model's predictions and provides confidence in its ability to identify novel functional proteins. The fact that ESM-2 naturally focuses on catalytically important regions without explicit training on protein function suggests that the model has learned fundamental principles of protein structure-function relationships from sequence data alone.
The development of the trans-cleavage activity prediction model addressed a critical challenge in protein characterization: the limited availability of experimental data for training machine learning models.
Using only 69 labeled Cas12a proteins, the researchers developed a LightGBM model that achieved 92.3% accuracy in predicting trans-cleavage activity. This success demonstrates the power of combining large pre-trained models with small-scale experimental data to predict specific protein functions.
Future Implications and Limitations
While this research represents a significant advance in AI-driven protein discovery, the authors acknowledge several limitations.
The model's dependence on training data means it may miss entirely novel protein architectures, and its high computational requirements limit accessibility for some researchers. The interpretability of transformer models, while improving, remains a challenge for understanding the biological principles underlying predictions.
The research opens numerous avenues for future investigation. The identified Cas12a variants require extensive characterization to fully understand their potential applications.
The broader approach could be applied to discover novel proteins in other families, potentially revolutionizing enzyme discovery and protein engineering. The integration of structural prediction with functional characterization provides a template for comprehensive protein characterization pipelines.
The ethical considerations surrounding CRISPR technology development also warrant attention. While these tools offer tremendous therapeutic potential, their misuse could pose biosecurity risks. The authors emphasize the need for stringent regulatory frameworks and ethical guidelines to ensure responsible application of these powerful technologies.
Conclusion
This groundbreaking research demonstrates the transformative potential of large language models in biological discovery. By combining ESM-2's protein understanding with careful experimental validation, the researchers have expanded the CRISPR-Cas12a toolkit and provided new insights into bacterial immune system evolution. The discovered variants offer immediate applications in gene editing and molecular diagnostics while opening new research directions in protein engineering and evolution.
The AIL-Scan approach represents a new paradigm for protein discovery that could accelerate the identification of novel enzymes, therapeutics, and biotechnological tools. As metagenomic databases continue to grow and computational methods improve, AI-driven approaches will likely become essential for mining the vast reservoir of undiscovered protein diversity in nature.
The success of this research underscores the importance of interdisciplinary collaboration between computational biology, structural biology, and experimental biochemistry. Future advances will likely require continued integration of these approaches to fully realize the potential of AI in biological discovery and application.
Definitions
CRISPR-Cas12a: A class 2 CRISPR system using a single, programmable endonuclease for RNA-guided DNA cleavage, distinct from Cas9 in its PAM requirements and cleavage patterns.
ESM (Evolutionary Scale Model): A transformer-based language model developed by Meta AI, trained on protein sequences to understand structure-function relationships at atomic resolution.
PAM (Protospacer Adjacent Motif): Short DNA sequences required by CRISPR-Cas systems for target recognition and cleavage initiation.
Trans-cleavage activity: The ability of activated CRISPR-Cas proteins to non-specifically cleave single-stranded DNA, used in diagnostic applications.
Metagenomics: The study of genetic material recovered directly from environmental samples, providing access to uncultured microbial diversity.
Integrase proteins (Cas1, Cas2, Cas4): Proteins responsible for incorporating new spacer sequences into CRISPR arrays during adaptive immune responses.
AlphaFold2: Google DeepMind's AI system for predicting protein structures from amino acid sequences with near-experimental accuracy (Jumper et al., 2021).
Cryo-EM: Cryogenic electron microscopy, a technique for determining high-resolution structures of biological molecules in their native states.


AI-Powered Discovery of Novel CRISPR-Cas12a Protein Clades Using Large Language Models
AI-Powered Discovery of Novel CRISPR-Cas12a Protein Clades Using Large Language Models