Alzheimer's disease (AD) is a devastating neurodegenerative condition, and while genetic factors like mutations in APP, PSEN, or MAPT and the APOE4 allele are known risk factors, they don't fully explain the widespread occurrence of late-onset AD (LOAD) in the general population.
Nearly everyone aged 65 or older develops some level of AD pathology, even without these known genetic predispositions. This gap in understanding highlights the critical need to identify other contributors to the disease.
Recent research has begun to shed light on the role of changes beyond the genetic sequence itself, specifically epigenomic changes and transcriptional dysregulation, in LOAD patients.
This article explores a groundbreaking study that identifies phosphoglycerate dehydrogenase (PHGDH), a protein previously known for its role in metabolism, as a key driver of amyloid pathology in AD through a previously unrecognized transcriptional regulatory function, independent of its enzymatic activity.
This discovery points towards novel therapeutic strategies for LOAD that move beyond targeting traditional genetic pathways.
Key Takeaways
- Elevated PHGDH could be an Early Alzheimer's Predictor: Increased expression levels of PHGDH are associated with the early stages of Alzheimer's disease and correlate with the severity of the disease.
- PHGDH Initiates Amyloid Pathology: High levels of PHGDH can trigger the development of amyloid pathology, a hallmark of Alzheimer's, even in the absence of common genetic risk factors like familial mutations or the APOE4 allele.
- Newly Identified Transcription Mechanism: The study reveals that PHGDH promotes amyloid pathology through its role in regulating gene transcription, a function independent of its established enzymatic activity in serine synthesis.
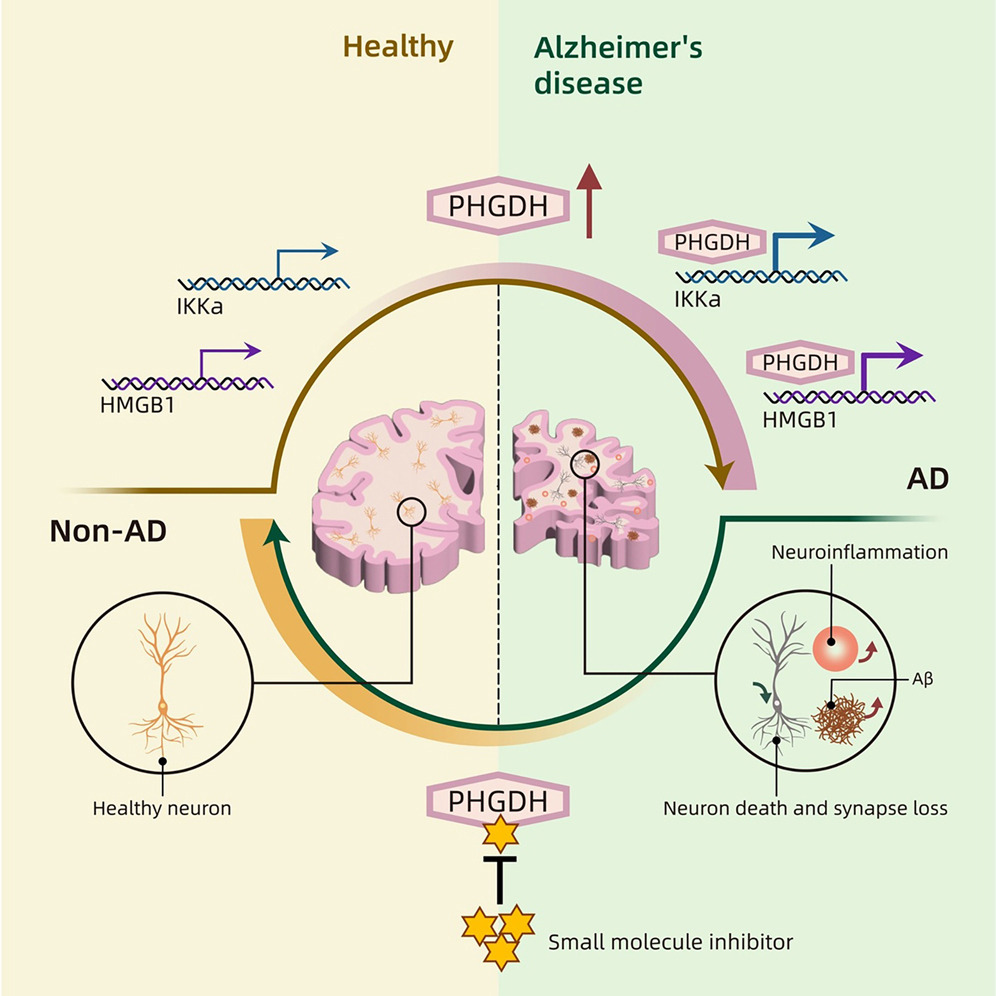
- Accelerates LOAD Pathology by Autophagy Supression: PHGDH in astrocytes promotes the transcription of inhibitor of nuclear factor kappa-B kinase subunit alpha (IKKα) and high-mobility group box 1 (HMGB1). This, in turn, suppresses autophagy (the cell's waste disposal system) and accelerates amyloid pathology.
- Potential Therapeutic Target: A blood-brain-barrier-permeable small molecule inhibitor (NCT-503) that targets PHGDH's transcriptional regulatory function has been shown to reduce Alzheimer's-related symptoms and pathology in mouse models and human brain organoids. This suggests a promising new therapeutic strategy focusing on transcriptional regulation rather than solely on familial mutations or established pathways.
- Possible Applications:
- Early Diagnosis: PHGDH levels, particularly its extracellular RNA (exRNA) in blood plasma, could serve as an early diagnostic biomarker for LOAD, even before clinical symptoms appear.
- Novel Drug Targets: Targeting the transcriptional regulatory function of PHGDH, or its downstream targets IKKα and HMGB1, could lead to new treatments for LOAD.
- Understanding Pathology Mechanisms: This research provides a deeper understanding of how LOAD can develop in the general population, particularly in individuals without known genetic predispositions
Overview
Late-onset Alzheimer's disease (LOAD) represents the vast majority of AD cases, yet its development is not solely explained by known genetic mutations associated with familial AD (FAD), such as mutations in APP, PSEN, or MAPT. Over half of LOAD patients do not carry the APOE4 risk allele.
This suggests that other factors contribute significantly to the disease process in the general population. While transcriptional dysregulation hasn't historically been considered a primary hallmark of AD, recent studies have revealed substantial epigenomic alterations in LOAD patients.
Phosphoglycerate dehydrogenase (PHGDH) has emerged as a prominent biomarker for AD, with its protein and RNA expression levels correlating with LOAD severity. Notably, elevated levels of PHGDH's extracellular RNA (exRNA) in blood plasma have been observed even before clinical diagnosis, suggesting its potential for early detection.
Unlike other biomarkers such as β-amyloid (Aβ) and phosphorylated Tau (p-Tau), which involve post-translationally modified proteins, both PHGDH protein and RNA expression levels correlate with LOAD. However, the question remained whether PHGDH was merely a marker or if it played a causal role in AD.
This research establishes a causal link, showing that overexpression of PHGDH promotes amyloid pathology, while under expression suppresses it.
This causal relationship was observed even in a human brain organoid model of LOAD (LOAD-BO) developed from embryonic stem cells lacking FAD, tauopathy, and APOE mutations, which displayed AD pathology when exposed to human blood serum.
PHGDH is known as a key enzyme in the synthesis of L-serine, an essential metabolite primarily produced in cells like astrocytes in the brain (a type of glial cell in the central nervous system (CNS) that play crucial roles in brain function, including supporting neurons, maintaining the blood-brain barrier, and regulating neurotransmitter levels). Disrupting this enzymatic function can affect L-serine supply and lead to severe developmental disorders
However, this study uncovered a previously uncharacterized function of PHGDH: transcriptional regulation. This means that PHGDH can directly influence the expression of other genes by interacting with DNA. The research suggests that this transcriptional regulatory activity, rather than its metabolic (enzymatic) function, is responsible for initiating early AD pathology.
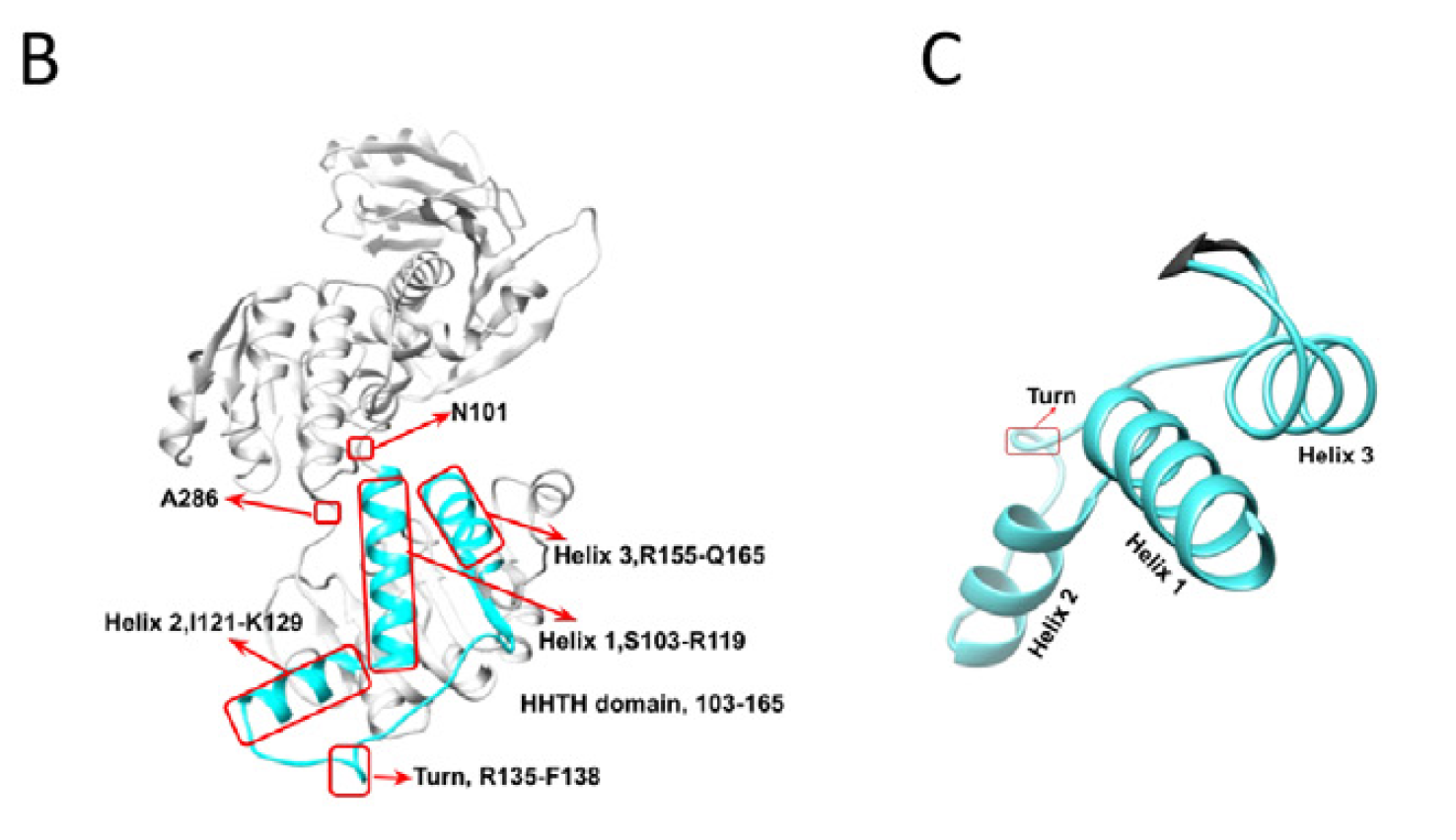
The research reveals that a portion of PHGDH localizes to the nucleus in astrocytes, where it can associate with chromatin, particularly in promoter regions of genes. This interaction is dependent on a specific structural domain within PHGDH called the helix-helix-turn-helix (HHTH) domain (identified using AlphaFold3), which shows similarity to DNA-binding motifs found in other transcription factors.
The study found that PHGDH achieves this by promoting the transcription of two specific genes in astrocytes: inhibitor of nuclear factor kappa-B kinase subunit alpha (IKKa) and high-mobility group box 1 (HMGB1).
IKKa is a crucial component of the NF-kB signaling pathway, known to be involved in inflammation and potentially promoting amyloid accumulation. While HMGB1 is released by stressed cells and can exacerbate inflammation, impairing the clearance of amyloid-beta (Aβ) and amplifying pathology.
These proteins are known to suppress autophagy (a cellular cleaning process) and activate inflammatory pathways (like NF-κB signaling), both of which are implicated in the progression of neurodegenerative diseases and can accelerate amyloid pathology.
By upregulating IKKa and HMGB1, astrocytic PHGDH stimulates downstream pathways like NF-kB and mTOR while suppressing autophagy (a natural cellular process where a cell breaks down and recycles damaged or unnecessary components like organelles and proteins), processes that are implicated in the acceleration of amyloid pathology.
Why it's Important
This research is highly significant for several reasons. Firstly, it addresses the critical question of how LOAD develops in individuals without classical genetic risk factors, a population that includes the vast majority of those affected.
By identifying PHGDH's transcriptional role as a causal mechanism, the study provides a new framework for understanding the onset and progression of LOAD in the general aging population. The identification of a transcriptional mechanism means that interventions could be designed to modulate gene expression rather than just metabolic pathways
Next, the discovery of PHGDH's non-enzymatic, transcriptional function as the pathological driver is a major paradigm shift. PHGDH has a known metabolic role in serine synthesis, and previous therapeutic approaches might have focused on inhibiting this function.
However, the study shows that simply inhibiting the enzyme's active site is not sufficient to prevent pathology. This highlights the need for new therapeutic strategies that specifically target the transcriptional regulatory activity.
The identification of IKKa and HMGB1 as key downstream targets of PHGDH provides specific molecular targets for intervention. The proposed PHGDH–IKKa–HMGB1 regulatory axis offers a potential new pathway to target therapeutically.
The study's success in using a blood-brain-barrier-permeable small-molecule inhibitor, NCT-503, to reduce amyloid pathology and improve behavioral deficits in mouse models is particularly promising.
NCT-503 is shown to interact with PHGDH's HHTH domain, crucial for its transcriptional function, rather than the enzymatic active site. This demonstrates the feasibility of targeting this specific mechanism.
Importantly, NCT-503's beneficial effects were independent of L-serine concentration and did not significantly affect brain L-serine levels in mice at the tested dose, mitigating concerns about disrupting the essential metabolic function of PHGDH. This opens up possibilities for developing drugs that can effectively treat AD pathology without causing metabolic side effects.
Finally, the observation that elevated PHGDH exRNA levels are detectable in blood plasma before clinical diagnosis reinforces its value as an early biomarker and suggests the potential for early diagnosis and intervention, which is crucial for effectively treating neurodegenerative diseases.
The findings are also notable because PHGDH expression doesn't correlate with the APOE4 genotype and its modulation affects Aβ and synapses without impacting p-tau levels, suggesting a distinct pathway contributing to neurodegeneration that is independent of two well-established AD factors.
Beyond Alzheimer's disease, the moonlighting function of PHGDH in transcriptional regulation could also provide insights into other conditions where PHGDH is implicated, such as certain developmental disorders (e.g., Neu-Laxova syndrome 1) and cancers.
The paper suggests that some mutations linked to severe developmental abnormalities, like R163Q, might exert their effects by disrupting PHGDH's transcriptional functions rather than solely its enzymatic activity, offering a new lens through which to view the pathology of these conditions.
In essence, this research broadens our understanding of AD pathogenesis, points to new diagnostic and therapeutic targets, and highlights the complex, multifaceted roles that proteins can play within a cell beyond their classically defined functions.
Summary of Results
The study employed a multi-faceted approach using mouse models and human brain organoids to investigate the role of PHGDH in AD pathology.
Initial observations in human data indicated that elevated PHGDH protein and RNA expression levels correlated with LOAD severity. Furthermore, extracellular RNA (exRNA) of PHGDH was increased in the blood plasma of LOAD patients prior to clinical diagnosis, suggesting its potential as an early diagnostic biomarker. Linkage studies had previously identified the PHGDH gene region as a strong LOAD linkage region.
To test causality, the researchers manipulated PHGDH expression in mouse models. Overexpression of human PHGDH in astrocytes of 3xTg-AD mice (a triple-transgenic model of FAD) promoted amyloid pathology.
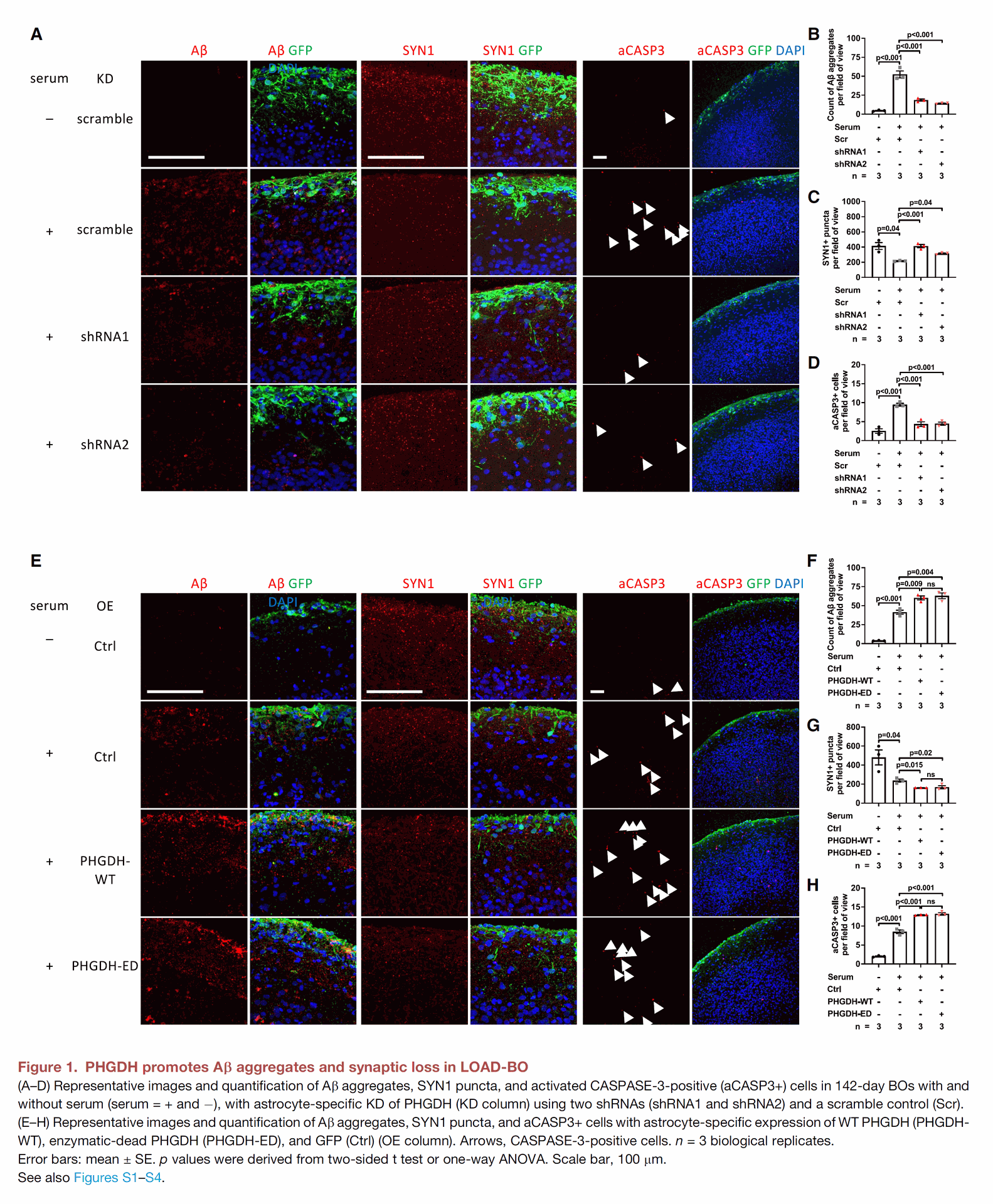
Figure 1 focuses on demonstrating the causal role of PHGDH expression levels in promoting or suppressing amyloid pathology in a model of late-onset Alzheimer's disease (LOAD) using human brain organoids.
- Panels A-D show that reducing (Knockdown or KD) the level of PHGDH in astrocytes within these organoids decreased the amount of amyloid-beta (Ab) aggregates, reduced synapse loss (measured by SYN1 puncta), and lowered cell death (indicated by activated CASPASE-3 positive cells). It's noted that reducing PHGDH did not affect p-Tau levels.
- Panels E-H show the opposite effect: increasing (Overexpression or OE) the level of PHGDH in astrocytes in these organoids made Ab accumulation, synapse loss, and cell death worse. Similar to the reduction experiments, PHGDH overexpression did not affect p-Tau levels. Importantly, expressing a version of PHGDH that cannot perform its usual enzymatic activity (PHGDH-ED) still exacerbated these AD-related changes just as much as the normal version (PHGDH-WT).
More critically, overexpression in wild-type mice (without FAD mutations) or double-transgenic mice with inducible astrocytic PHGDH expression also led to elevated Aβ without requiring disease-causing mutations or the APOE4 allele.
While PHGDH overexpression alone increased Aβ, it was noted that it wasn't sufficient to fully replicate all aspects of LOAD pathology. Conversely, underexpression of PHGDH suppressed amyloid pathology.
The researchers then utilized human brain organoids (BOs) derived from human embryonic stem cells lacking FAD, MAPT, or APOE mutations. When exposed to human blood serum to model LOAD, these BOs developed significant Aβ aggregates and other LOAD-associated pathologies, accompanied by elevated astrocytic PHGDH expression.
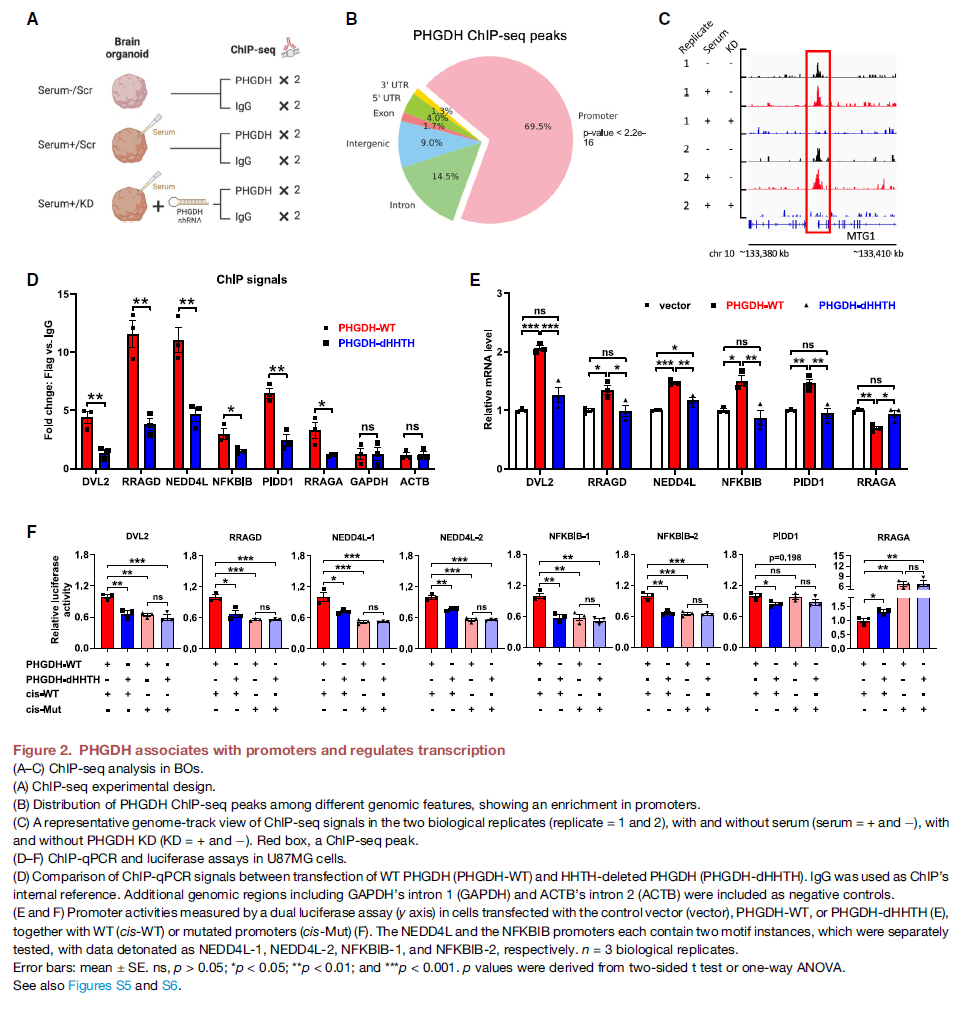
Figure 2 explores how PHGDH works at a molecular level, showing that it can bind to DNA and regulate gene expression.
- Panels A-C present data from ChIP-seq experiments, which identify where a protein binds to DNA in the genome. These results show that PHGDH primarily binds to gene promoter regions. Further analysis suggests these binding sites are linked to genes involved in neurodegenerative diseases.
- Panels D-F investigate the mechanism of PHGDH's DNA binding and regulatory function using cell experiments. They demonstrate that PHGDH's ability to bind to chromatin (DNA) and control gene activity depends on a specific structural part called the HHTH domain. These panels also reinforce that PHGDH's impact on gene expression does not require its enzymatic activity.
Astrocyte-specific knockdown (KD) of PHGDH using shRNAs in serum-exposed BOs significantly reduced the number and coverage area of Aβ aggregates, increased synaptic marker (SYN1) puncta, and decreased apoptotic cells (aCASP3+), while not affecting p-Tau levels.
This contrasts with findings about p-Tau being a key driver of neurodegeneration. Conversely, astrocyte-specific PHGDH overexpression in BOs exacerbated these pathological changes.
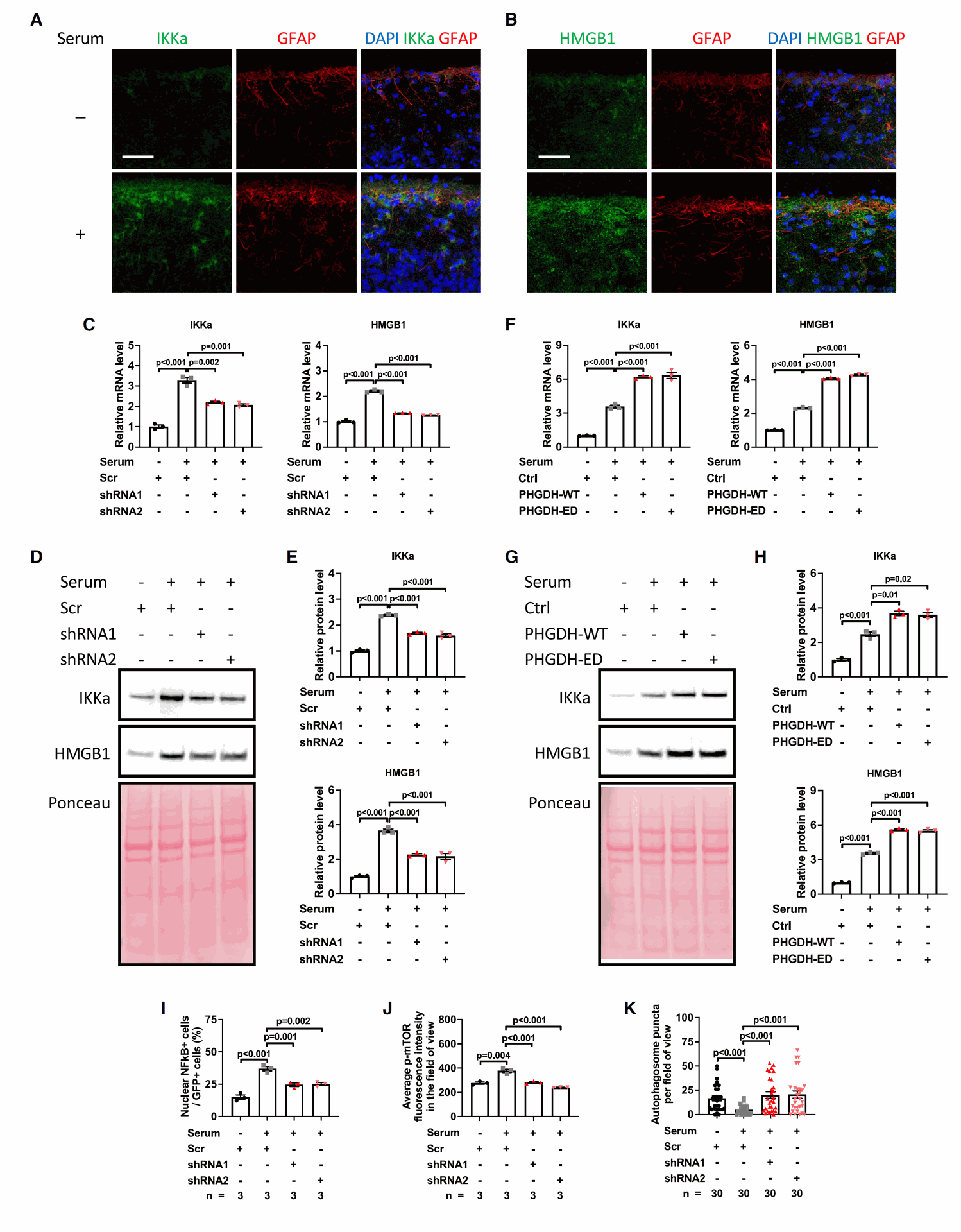

Figure 3 identifies and shows the regulation of specific target genes of PHGDH, namely IKKa and HMGB1.
- Panels A and B show images confirming the presence of IKKa and HMGB1 in astrocytes in brain organoids.
- Panels C-E show that when PHGDH is reduced (KD) in astrocytes in the LOAD model organoids, the levels of both IKKa and HMGB1 messenger RNA and protein decrease.
- Panels F-H show that when PHGDH is increased (OE) in astrocytes, the levels of IKKa and HMGB1 increase. This increase in IKKa and HMGB1 expression is independent of PHGDH's enzymatic activity.
- Panels I-K demonstrate that the increase in activity of NF-kB and mTOR, along with decreased autophagy observed in astrocytes in the LOAD model organoids, is dependent on the expression of astrocytic PHGDH.
A crucial experiment involved overexpressing an enzymatically inactive form of PHGDH (PHGDH-ED) in BOs. This mutant, with its active site modified, still exacerbated Aβ accumulation, synaptic loss, and apoptosis, with effects similar to wild-type (WT) PHGDH overexpression. This strongly indicated that PHGDH's role in AD pathology is independent of its serine synthesis enzymatic activity.
Investigating the mechanism further, the study found that PHGDH localizes to the nucleus in astrocytes and neural stem cells. Chromatin immunoprecipitation sequencing (ChIP-seq), a technique used to identify DNA regions that a specific protein binds to, revealed that PHGDH associates with chromatin, showing a significant enrichment in promoter regions.
This supports its role as a transcriptional regulator. Motif analysis identified an enriched DNA sequence in PHGDH binding sites with a core sequence similar to that recognized by the PBX family of TALE-homeodomain proteins, suggesting a potential mechanism for DNA binding.
Single-cell RNA sequencing (snRNA-seq) of the BOs allowed the researchers to identify cell-type specific gene expression changes. Through a prioritization process involving ChIP-seq data and expression changes, IKKa and HMGB1 were identified as key transcriptional targets of PHGDH in astrocytes.
Astrocyte-specific KD of PHGDH significantly reduced IKKa and HMGB1 mRNA and protein levels, while overexpression (including the enzymatically inactive form) significantly increased them. This confirmed that PHGDH regulates these genes transcriptionally and independently of its enzymatic activity.
Further experiments showed that in LOAD-BOs, astrocytic PHGDH expression increased nuclear-localized NF-kB and phosphorylated mTOR activity while decreasing autophagy, changes that were dependent on PHGDH expression.
Simultaneous knockdown of both IKKa and HMGB1 in BOs with PHGDH overexpression effectively rescued the AD-related pathologies induced by PHGDH, including reducing Aβ aggregates, increasing SYN1, and decreasing aCASP3+ cells. This demonstrates that IKKa and HMGB1 mediate PHGDH's pathological effects.
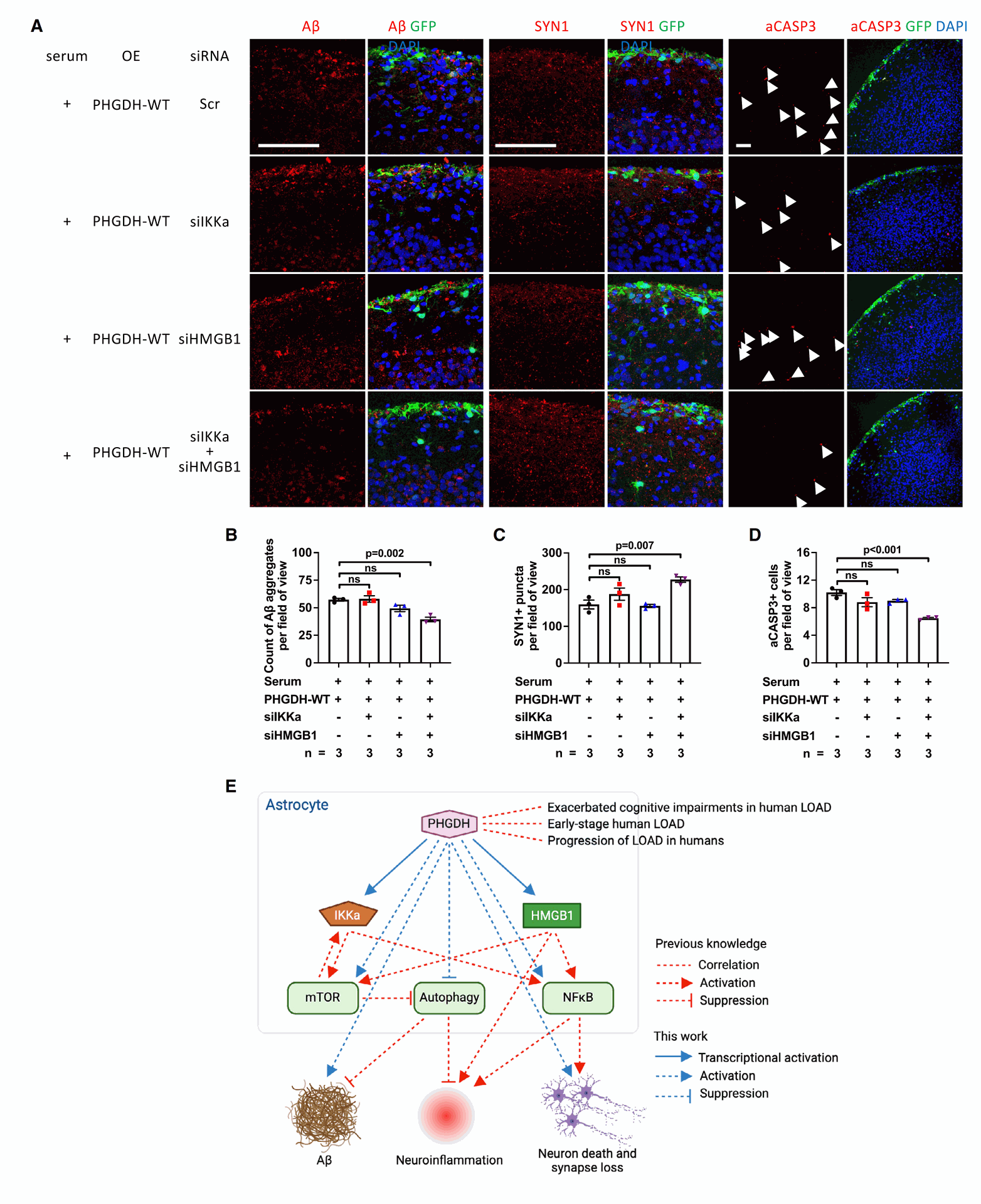

Figure 4 tests whether the PHGDH targets, IKKa and HMGB1, are necessary for PHGDH to cause AD-related pathology.
- Panels A-D show experiments in brain organoids where PHGDH is overexpressed (to induce pathology), and then the levels of IKKa or HMGB1, or both, are lowered using siRNAs. The results indicate that reducing either IKKa or HMGB1 alone did not significantly improve the AD-related problems caused by high PHGDH levels. However, reducing both IKKa and HMGB1 simultaneously significantly reduced Ab aggregates, increased synapse markers, and decreased cell death, effectively reversing the pathological effects of PHGDH overexpression.
- Panel E presents a model that summarizes the proposed pathway: PHGDH activation in astrocytes stimulates IKKa and HMGB1, which then activate NF-kB and mTOR pathways and suppress autophagy, contributing to LOAD development.
Finally, the researchers tested a blood-brain-barrier (BBB)-permeable small-molecule inhibitor, NCT-503, previously shown to inhibit PHGDH. Molecular docking (AlphaFold) suggested NCT-503 interacts with residues within or near PHGDH's HHTH DNA-binding domain, unlike control compounds.
Treatment with NCT-503 significantly reduced Aβ aggregates, increased SYN1, and reduced apoptotic cells in LOAD-BOs, while a compound targeting enzymatic activity (IPA) or supplementing L-serine did not.
In mouse models, NCT-503 administration reduced Aβ plaques in 5XFAD mice and APP-KI mice. Importantly, at the tested dose, NCT-503 did not significantly affect brain L-serine levels in mice but did reduce IKKa and HMGB1 mRNA and protein levels supporting its mechanism of action on transcriptional regulation rather than enzymatic activity. NCT-503 treatment also improved behavioral deficits (anxiety-like behavior and spatial memory) in APP-KI mice, correlating with reduced amyloid pathology.
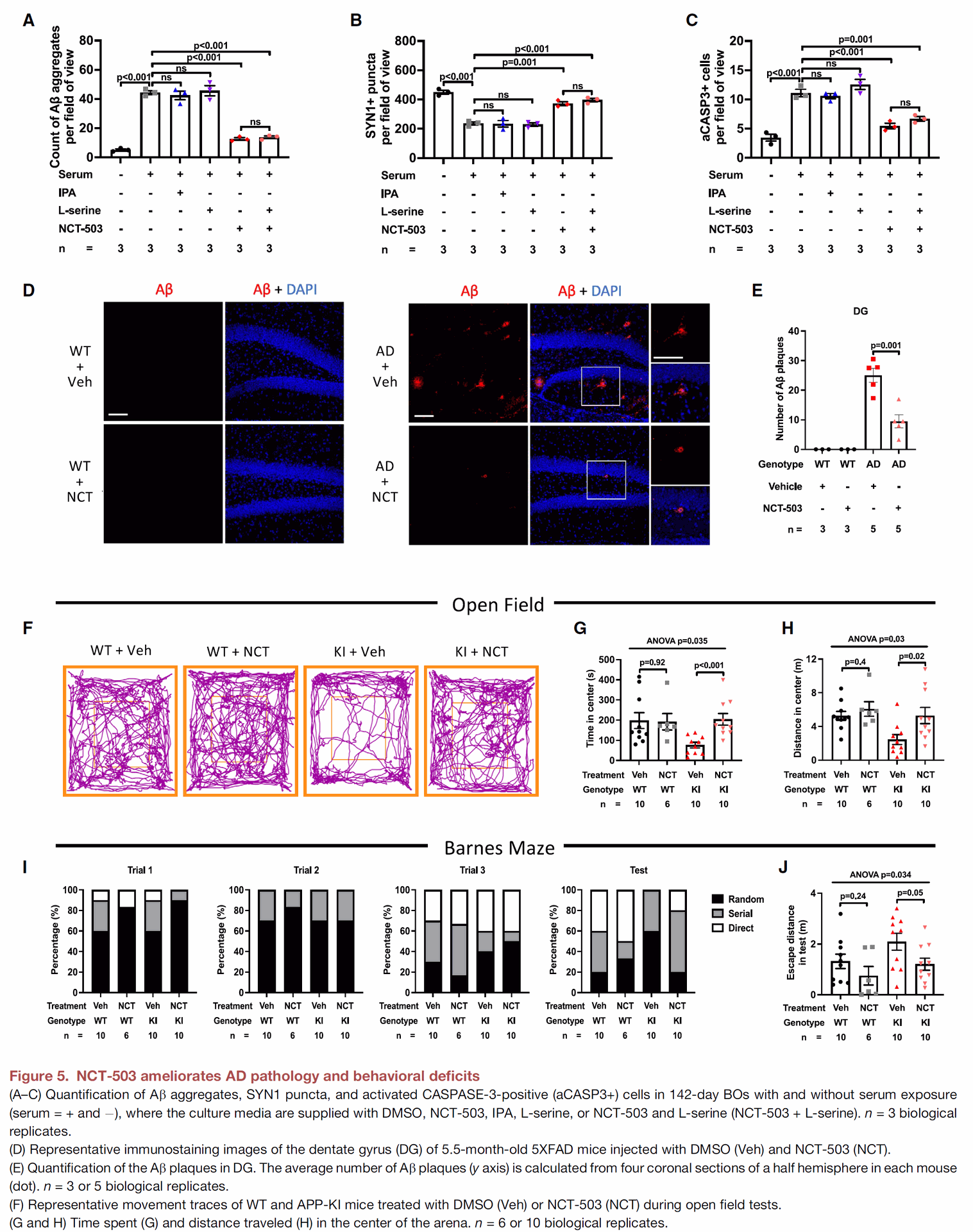
Figure 5 tests a potential small-molecule inhibitor, NCT-503, designed to target PHGDH's transcriptional function.
- Panels A-C show that treatment with NCT-503 improves the AD-related problems in brain organoids, reducing Ab aggregates, improving synapse markers, and decreasing cell death in the LOAD model. A related compound, IPA, did not have these beneficial effects. The positive effects of NCT-503 were found to be independent of L-serine concentration. These findings support that NCT-503 works by inhibiting PHGDH's transcriptional function, not its enzymatic one.
- Panels D and E show that administering NCT-503 to 5XFAD mice, a mouse model of amyloid pathology in AD, significantly reduces the number and size of amyloid plaques in their brains.
- Panels F-J demonstrate that NCT-503 treatment in APP-KI mice, another AD mouse model, led to reduced anxiety-like behavior and improved spatial memory. In these mice, NCT-503 treatment also resulted in lower levels of IKKa and HMGB1 in the brain, consistent with NCT-503 targeting PHGDH's transcriptional regulation.
The authors also include detailed methods (STAR Methods section ) and discuss limitations, such as the inhibitor studies being conducted in FAD mice while causal links were shown in LOAD models, and the need for broader behavioral assessments.
Conclusion
This research fundamentally changes our understanding of how Alzheimer's disease pathology can develop, particularly in the vast population affected by late-onset AD without classical genetic mutations. It firmly establishes PHGDH, a protein known for its metabolic role, as a causal driver of amyloid pathology through a novel, enzymatic-independent transcriptional regulatory function.
The study provides compelling evidence that PHGDH localizes to the nucleus and associates with gene promoters, transcriptionally activating genes like IKKa and HMGB1 in astrocytes. This activation subsequently suppresses autophagy and accelerates amyloid accumulation. The demonstration that simultaneously targeting IKKa and HMGB1 can rescue PHGDH-induced pathologies highlights their critical role in this pathway.
Perhaps the most exciting finding is the validation of a BBB-permeable small-molecule inhibitor, NCT-503, that targets PHGDH's transcriptional function. NCT-503 successfully reduced amyloid pathology and improved behavioral deficits in relevant mouse models without disrupting essential serine metabolism. This underscores the therapeutic potential of targeting this newly identified PHGDH transcriptional pathway.
These insights offer a promising avenue for developing new treatments for LOAD, potentially benefiting the majority of AD patients who do not carry familial AD mutations or the APOE4 allele. The potential for early diagnosis using PHGDH exRNA levels in blood further enhances the significance of this work. While the study notes limitations, such as the need for further behavioral assessments and exploring alternative drug formulations, the identification of the PHGDH–IKKa–HMGB1 axis and the validation of a transcriptional inhibitor like NCT-503 represent a significant step forward in the fight against Alzheimer's disease.


Unraveling a New Culprit in Alzheimer's Disease: PHGDH Transcriptional Regulation
Transcriptional regulation by PHGDH drives amyloid pathology in Alzheimer’s disease