The exponential growth of make-on-demand chemical libraries offers unprecedented opportunities for early drug discovery. However, screening these multi-billion-scale (potentially multi-trillion soon) libraries remains computationally challenging, even for fastest structure-based docking methods.
The authors explore a strategy that combines machine learning and molecular docking to enable rapid virtual screening of databases. This machine learning-guided docking approach significantly reduces computational costs while enabling efficient virtual screening.
Full Paper on Nature CP Framework Githhub Repo
Key Takeaways
- A novel workflow integrating machine learning and molecular docking screens databases of billions of compounds efficiently.
- The conformal prediction framework is used to make selections from the multi-billion-scale library, drastically reducing the number of compounds to be scored by the docking algorithm.
- The CatBoost classifier achieves an optimal balance between speed and accuracy, guiding the selection of promising compounds.
- The protocol reduces the computational cost of structure-based virtual screening by more than 1,000-fold.
- Experimental validation confirmed the discovery of ligands targeting G protein-coupled receptors (GPCRs), demonstrating the potential of this approach for multi-target drug discovery.
- The method facilitates the discovery of compounds with activity at multiple drug targets, which is crucial for treating complex diseases like Parkinson’s.
- Application to a library of 3.5 billion compounds demonstrated that molecules with substantially improved docking scores can be identified by machine learning, enabling efficient virtual screening of the largest commercial chemical libraries available.
Overview
Traditional virtual screening methods struggle to evaluate ultra-large chemical libraries due to computational constraints. This research introduces a hybrid strategy combining machine learning with molecular docking, significantly accelerating the screening process.
The method involved training a classifier on a dataset of 1 million compounds, using a novel conformal prediction framework to identify top candidates from multi-billion-scale libraries. This approach enables the selection of promising molecules with reduced computational overhead.
Background
The number of possible drug-like molecules is estimated to be more than 1060, exceeding the size of chemical libraries evaluated in early drug discovery by many orders of magnitude.
Currently, only about 13 million compounds are available in-stock from chemical suppliers, highlighting the limited coverage of chemical space. Advances in synthetic organic chemistry have expanded compound collections, with make-on-demand libraries containing over 70 billion readily available molecules. These libraries offer diverse scaffolds, presenting a major opportunity for drug discovery, but identifying relevant compounds in this vast chemical space remains challenging.
Recent structure-based virtual screens of ultra-large libraries have identified ligands for important therapeutic targets, demonstrating that expanding the coverage of chemical space can accelerate early drug discovery.
However, screening these massive libraries is currently computationally intensive, and the make-on-demand databases will only continue to grow, potentially reaching trillions of compounds. This growth necessitates more efficient virtual screening approaches to evaluate these vast chemical libraries.
Machine Learning in Drug Discovery
Recent advancements in artificial intelligence have renewed interest in using quantitative structure-activity relationship (QSAR) models in drug discovery. QSAR models predict both on- and off-target activities, as well as physicochemical and pharmacokinetic properties.
By representing compounds using molecular descriptors, machine learning methods can rapidly evaluate large compound databases. Traditionally, QSAR models have been trained on experimental data, but there is increasing interest in predicting which compounds in make-on-demand libraries are likely to receive favorable scores from computationally expensive virtual screening methods.
Conformal prediction (CP) is a machine learning quantitative structure–activity relationship (QSAR) method in which an ensemble of models is used to classify molecules present in an objective set. It provides class-specific confidence levels, allowing compounds to be categorized into virtual active, virtual inactive, both (virtual active or inactive), and null (no class assignment) sets based on a selected significance level.
This framework enables control over the error rate of the predictions and is well-suited for handling inherently imbalanced datasets, such as those in virtual screening applications.
This combination of machine learning and molecular docking screening has the potential to enable virtual screens of multi-billion-scale compound libraries at a modest computational cost.
Why It’s Important
The research demonstrates that combining conformal prediction with machine learning can significantly reduce the computational cost of screening ultra-large chemical libraries. This approach enables the discovery of ligands with complex properties, which may be difficult to find in smaller libraries. The method's ability to identify compounds with multi-target activity tailored for therapeutic effect highlights its potential to accelerate drug discovery for complex diseases.
The study's workflow can be applied to identify starting points for the development of drugs with multi-target profiles tailored for treating complex diseases. This is particularly relevant for conditions like Parkinson's disease, where modulating multiple targets is necessary for effective treatment. The author's identification of a dual-target ligand for the A2A adenosine and D2 dopamine receptors demonstrates the practical application of this approach in drug discovery.
- Scalability: The proposed method addresses the scalability limitations of conventional virtual screening, making it feasible to explore vast chemical libraries.
- Drug Discovery Acceleration: By reducing the need for explicit docking, the approach expedites the identification of potential drug candidates.
- Multi-Target Ligand Discovery: The method facilitates the discovery of compounds with activity at multiple drug targets, which is crucial for treating complex diseases like Parkinson’s.
- Cost Efficiency: The workflow achieves a significant reduction in computational resource requirements, making large-scale virtual screening more accessible.
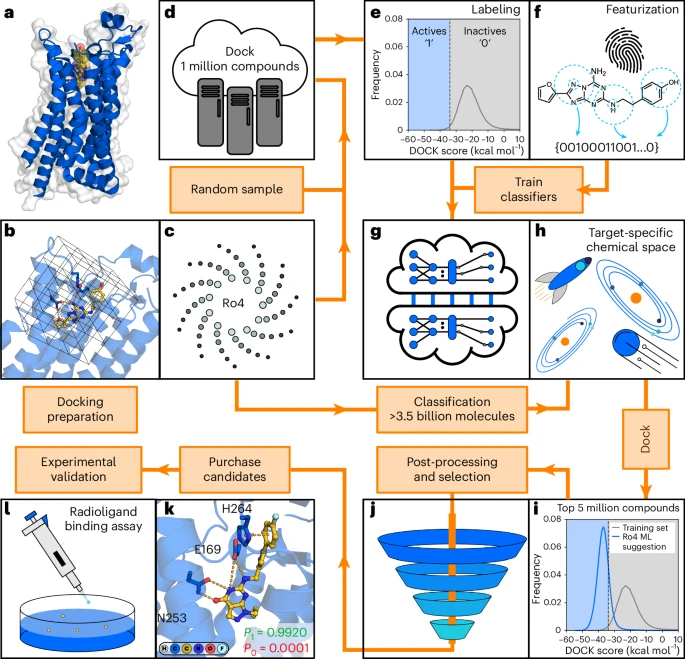
a, Selection of a target protein for virtual screening. b, Model representation of protein binding site for molecular docking calculations. c, A subset from an ultralarge (Ro4, rule-of-four) chemical library is extracted and prepared for docking screens. d, Docking scores for compounds in the training set are generated. e, A docking score threshold splits the training set into virtual actives (1 class) and inactives (0 class). f, Molecules are represented by descriptors (for example, fingerprints). g, Machine learning models are trained to distinguish virtual actives from inactives. h, The trained models are used to identify a subset of predicted virtual actives in the ultralarge library. i, A set of compounds is selected for docking to the target. Rule-of-four molecules suggested by machine learning (Ro4 ML) have an improved docking score distribution compared to random molecules in the training set. j, Post-processing of docking results and selection of compounds. k, Compounds are selected based on visual inspection. l, Experimental evaluation of synthesized compounds.
Conformal Prediction (CP) Framework
The authors introduce the CP framework an ultrafast machine learning technique that provides reliable confidence estimates for classification predictions. Unlike traditional models that output a single prediction, CP assigns a confidence level to each classification, allowing the user to control the error rate.
The CP framework can be applied to any machine learning classifier and allows the user to control the error rate of the predictions. Mondrian conformal predictors provide class-specific confidence levels that ensure validity for both the majority and minority classes.
It is particularly useful in handling imbalanced datasets, such as virtual screening, where only a small subset of compounds are expected to be active. The CP framework ensures that predictions remain valid across both majority and minority classes, making it an effective tool for filtering large chemical libraries efficiently predicting pharmacokinetic properties and bioactivity.
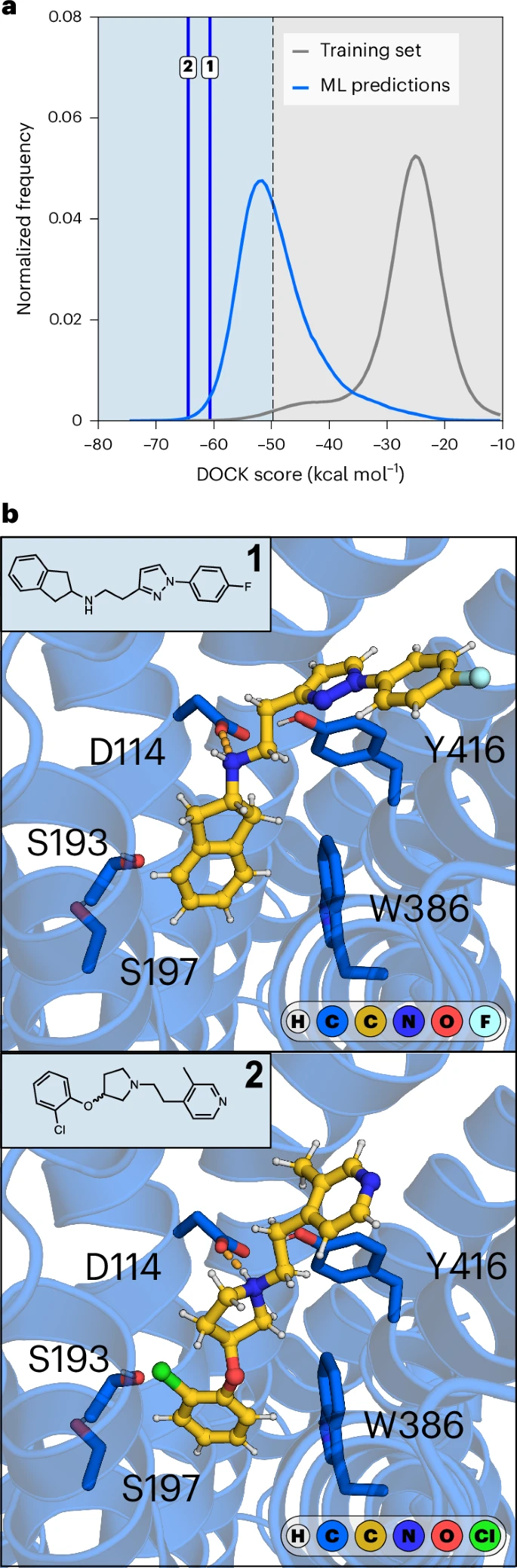
The machine learning-accelerated workflow was used to predict ligands of the D2R in a database of 3.5 billion compounds. a, Normalized frequency distributions of D2R docking scores. The docking score distribution of the training set is shown in gray. The docking score distribution of 5 million molecules selected based on the P value difference (P1 − P0) is shown in blue. Vertical blue lines indicate the docking scores of the experimentally verified ligands, compounds 1 and 2. b, Chemical structures and predicted binding modes of experimentally verified ligands (compounds 1 and 2, represented as gold sticks) of D2R (represented as a blue cartoon with side chains of key residues as sticks). Key hydrogen bonds (ionic interaction) are indicated by orange dashed lines.
Summary of Results
The study conducted molecular docking screens against eight therapeutically relevant proteins to evaluate the CP workflow. The results showed that the CatBoost classifier trained on Morgan2 fingerprints achieved the best average precision and had comparable or slightly better significance and sensitivity values compared with other combinations.
This configuration required the least computational resources, both in the training of the classifier, predictions for the test set, and storage of molecular descriptors.
The optimized workflow was demonstrated to identify more than 90% of the very top-scoring molecules in a library with 0.2 billion compounds, which only required docking of 3-5% of this set. Application to a library with over 3.5 billion compounds showed that molecules with substantially improved docking scores can be identified by machine learning, enabling efficient virtual screening of the largest commercial chemical libraries currently available.
The workflow achieved a 568-fold reduction of compute cost compared with explicit docking of the 3.5 billion compounds. The docking score distribution of the 5 million prioritized compounds was substantially shifted toward more favorable energies, with a large fraction of the predicted compounds having a docking score better than the energy threshold used for labeling of the training set.
- Development of the Workflow:
- A conformal prediction framework was applied to a machine learning classifier trained on docking data from 1 million compounds.
- The model was tested on 3.5 billion compounds, reducing the number of compounds requiring explicit docking by three orders of magnitude.
- Benchmarking of Conformal Predictors:
- The approach was validated against eight therapeutically relevant protein targets.
- The CatBoost classifier with Morgan2 fingerprints performed best in precision and efficiency.
- Achieved a 568-fold reduction of compute cost
- Application to GPCR Ligand Discovery:
- The method successfully identified ligands for the A2A adenosine receptor (A2AR) and D2 dopamine receptor (D2R).
- Experimental validation confirmed the activity of selected compounds in radioligand binding assays.
- Multi-Target Ligand Identification:
- The workflow was used to find compounds targeting both A2AR and D2R, relevant for Parkinson’s disease treatment.
- A dual-target ligand was identified, supporting the potential of this approach for polypharmacology.
Conclusion
This study demonstrates that machine learning-guided docking significantly improves the efficiency of virtual screening in ultra-large chemical libraries. By integrating conformal prediction with docking, the method reduces computational costs while maintaining accuracy.
Experimental validation underscores its practical utility in drug discovery, particularly for multi-target ligand identification. This approach has the potential to revolutionize the way virtual screening is conducted, paving the way for faster and more cost-effective drug discovery.

Rapid Traversal of Vast Chemical Space Using Machine Learning-guided Docking Screens