This research introduces a multifidelity Bayesian optimization (MF-BO) approach to accelerate the automated discovery of drug molecules by effectively leveraging experimental data of varying costs and accuracy.
Bayesian optimization is an approach to optimizing objective functions that take a long time (minutes or hours) to evaluate. It is a clever way to find the best settings (or inputs) for a function, especially when evaluating that function is time-consuming or expensive.
The study demonstrates that MF-BO can outperform traditional experimental funnels and single-fidelity Bayesian optimization by intelligently balancing the exploration of a chemical search space with the exploitation of promising candidates using different levels of experimental fidelity.
The authors addresses the challenge of efficiently identifying new drug molecules by combining the strengths of experimental funnels, which use sequential screenings of increasing fidelity, with Bayesian optimization, an iterative method for finding optimal solutions with fewer experiments.
By integrating multifidelity data into Bayesian optimization, this work aims to optimize the selection of experiments based on their cost and the quality of the data they provide, ultimately accelerating drug discovery.
Key Takeaways
- Multifidelity Bayesian optimization (MF-BO) significantly accelerates the discovery of drug molecules compared to traditional experimental funnels, transfer learning approaches, and Bayesian optimization using only high-fidelity data.
- MF-BO strategically allocates experimental budget across different fidelities (low, medium, and high) to efficiently explore the chemical search space and identify promising candidates.
- The effectiveness of MF-BO is dependent on the diversity of the chemical search space and the correlation between the different experimental fidelities. It excels in diverse spaces with weak-to-moderate correlation.
- The MF-BO algorithm was successfully integrated into an autonomous molecular discovery platform to prospectively identify new histone deacetylase inhibitors (HDACIs).
- The autonomous platform automatically performed docking simulations (low fidelity), single-point percent inhibition assays (medium fidelity), and facilitated manual dose-response curve measurements (high fidelity).
- The study identified several new HDACIs with submicromolar inhibition that lack problematic hydroxamate moieties found in many current inhibitors.
- The platform showcases the potential for fully automated and iterative drug discovery, integrating molecular design, synthesis, and testing guided by MF-BO.
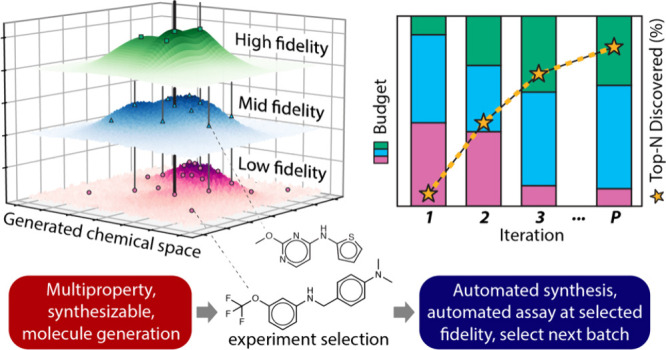
Overview
Traditional small molecule pharmaceutical discovery often employs an experimental funnel approach, which involves screening large libraries of molecules with rapid, low-cost assays, followed by progressively more accurate but expensive assays on a smaller subset of molecules.
This approach aims to balance the scale of screening with the accuracy of the results. In contrast, iterative design of experiments, such as Bayesian optimization (BO), uses feedback from each experimental iteration to optimally select the next set of experiments, often requiring fewer compounds to be screened to find high-performing molecules. However, conventional BO typically utilizes only a single level of experimental fidelity, often the most expensive one.
This research proposes and validates multifidelity Bayesian optimization (MF-BO), which integrates the concept of using multiple experimental fidelities from the experimental funnel with the iterative nature of Bayesian optimization.
The core idea is to leverage low-cost, low-fidelity experiments to efficiently explore a large chemical space and learn the landscape of potential drug candidates, while using more expensive, high-fidelity experiments to accurately evaluate the most promising ones. This allows for a more informed allocation of experimental resources, potentially leading to faster identification of optimal bioactive targets.
The authors highlight that the development of MF-BO in molecular discovery has been limited due to challenges such as unknown correlations between experiment levels and uncertainty about their costs. Previous advancements in multifidelity optimization have primarily occurred in computational settings, like finite element models, where costs and error bounds are well-defined.
This work extends existing multifidelity optimization heuristics, like targeted variance reduction (TVR) developed for simulations, to the context of molecular discovery with batched assays and discrete search spaces, where the correlation between fidelities is unknown and varies across the chemical space.
To evaluate the effectiveness of MF-BO, the researchers first applied the algorithm to five precompiled data sets from ChEMBL, comparing its performance to an experimental funnel (EF) approach, transfer learning with low-fidelity data (TL), and Bayesian optimization using only high-fidelity data (BO). The performance was measured by the rate of (re)discovery of top-performing molecules within a given budget.
Subsequently, the MF-BO algorithm was integrated into an autonomous molecular discovery platform to conduct a prospective search for new histone deacetylase inhibitors (HDACIs). They are a class of drugs that inhibit the activity of histone deacetylases used to treate cancer, neurological issues and heart disease.
This platform combines a genetic algorithm for generating diverse and synthesizable candidate molecules, the MF-BO algorithm for selecting candidates and their evaluation fidelity, and computer-aided synthesis planning (CASP) tools for automated synthesis. The physical platform includes robotic systems for liquid handling, synthesis, purification (HPLC-MS), and automated biochemical assays.
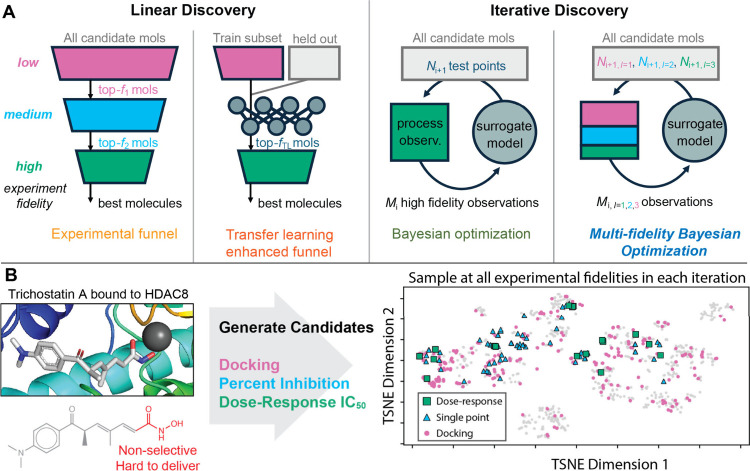
(A) A comparison of different types of design of experiments in small molecule drug discovery, divided into linear designs and iterative designs. (B) Application of MF-BO to the prospective discovery of new HDACIs. Shown at left is the crystal structure of trichostatin A bound to human HDAC8. The hydroxamic acid moiety (red) makes current HDACIs hard to deliver and dose safely. Generated molecules can be tested at three levels: docking, which is weakly correlated to potency; single-point percent inhibition, which is moderately correlated to potency; and dose–response measurement, which is a direct measurement of potency. The t-distributed stochastic neighbor embedding (TSNE) plot shows the selection of experiments of different fidelities over the generated chemical space in a single experimental iteration.
Figure 1 above provides a visual comparison of different design of experiments in small molecule drug discovery, contrasting linear designs (like experimental funnels) with iterative designs (like Bayesian optimization and MF-BO).
Figure 1B (above) illustrates the application of MF-BO to HDACI discovery, showing the crystal structure of trichostatin A bound to HDAC8 and the three levels of experimental fidelity used: docking (low), single-point percent inhibition (medium), and dose-response measurement (high). The t-distributed stochastic neighbor embedding (TSNE) plot in Figure 1B demonstrates the selection of experiments of different fidelities across the generated chemical space in a single iteration.
Why it’s Important
The development of MF-BO and its integration into an autonomous platform holds significant importance to the field of drug discovery for several reasons:
- Accelerated Drug Discovery By intelligently utilizing data from experiments of varying fidelities, MF-BO can potentially identify promising drug candidates much faster than traditional methods that rely on a linear progression through high-throughput screening and sequential assays. The iterative nature allows for learning from each experiment and focusing resources on the most promising areas of the chemical space.
- Resource Efficiency MF-BO can lead to more efficient use of experimental resources by prioritizing low-cost experiments for initial screening and exploration, reserving expensive high-fidelity assays for a smaller, more focused set of candidates. This is particularly crucial in drug discovery, where experimental costs can be substantial.
- Overcoming Limitations of Single-Fidelity Approaches Traditional Bayesian optimization often relies solely on high-fidelity data, which can be expensive and limit the number of molecules that can be explored. MF-BO overcomes this by incorporating information from lower fidelity experiments, enabling a broader exploration of the chemical space within a given budget.
- Autonomous Drug Discovery The integration of MF-BO with an automated platform demonstrates the potential for a fully autonomous drug discovery pipeline. This could significantly reduce the need for manual intervention, accelerate the pace of research, and enable the exploration of larger and more complex chemical spaces. The modular design of the platform, as mentioned in the paper and visually represented in Figure 3C, ensures robustness and the ability to continue operations even if one instrument experiences an error (as seen with the HPLC failure in Figure 4A).
- Addressing Challenging Drug Targets The successful identification of novel HDACIs without hydroxamate moieties highlights the potential of this approach to tackle challenging drug targets and design molecules with improved properties (e.g., delivery and safety).
Beyond drug discovery, the principles of MF-BO could be applied to other domains where iterative optimization using experiments or simulations of varying fidelities is common, such as materials science, chemical engineering, and robotics.
The ability to efficiently explore a design space by strategically using different levels of information could lead to faster and more cost-effective optimization in these fields as well.
Summary of Results
The researchers presented results in two main parts: evaluation of the MF-BO algorithm on literature data sets and its prospective application to discover HDACIs using the autonomous platform.
Part 1: Evaluation of MF-BO
- The MF-BO algorithm, using a Gaussian process (GP) surrogate model with Morgan fingerprints (a type of molecular fingerprint that represent a molecule's structure as a bit vector) and a Tanimoto kernel (a common similarity measure commonly used to compare molecular fingerprints) , outperformed experimental funnel (EF), transfer learning (TL), and single-fidelity Bayesian optimization (BO) on five ChEMBL data sets (complement factor D, CXCR4, PARP1, HIF-PH, and NR1A2).
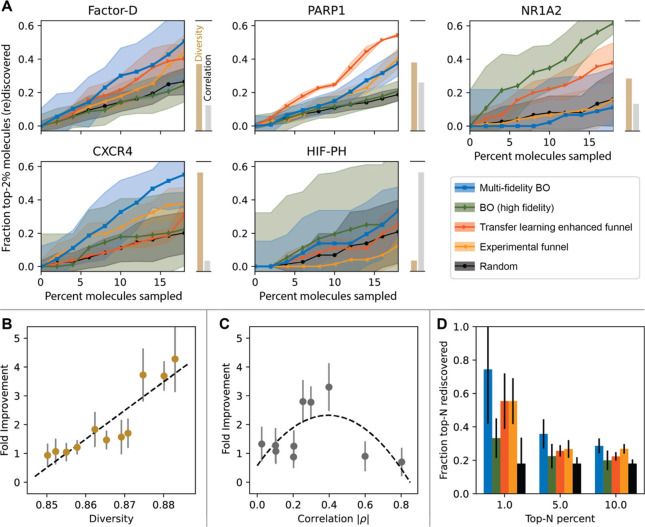
Figure 2 (A) Comparison of MF-BO against other designs of experiments. Markers indicate the mean fraction of top-2% molecules found after sampling the search space in discrete 2% batches. Shaded regions are the standard deviation from 6 repeats of each experiment. Blue ■ - MF-BO, green ◆ - BO, red ▶ - transfer learning, orange ◀ - experimental funnel, black ● - random selection. Gold and silver bars represent scaled diversity and correlation, respectively, within target data sets. (B, C) Enhancement of MF-BO over BO using structured data sets sampled from acetylcholinesterase inhibitors to probe the effect of diversity (B, gold) and fidelity correlation (C, silver) on MF-BO usefulness. Dashed curves are to guide the eye. (D) Assessment of different designs of experiments when rediscovering top-N% molecules in the Factor-D data set after sampling 18% of the data set. Colors are the same as in (A) and error bars represent the standard deviation of 6 repeats.
- Figure 2A visually compares the performance of MF-BO against these other methods by showing the mean fraction of top-2% molecules found after sampling in discrete 2% batches. The shaded regions represent the standard deviation from multiple repeats, highlighting the robustness of MF-BO in certain cases.
- The study found that MF-BO showed substantial acceleration (around 30% improvement for Factor-D) in drug targets with diverse search spaces and weak-to-moderate correlation between experimental fidelities. This is illustrated in Figure 2A by the gold (diversity) and silver (correlation) bars alongside the performance plots for each target.
- Figure 2B and 2C further investigated the impact of diversity and fidelity correlation using structured subsets of acetylcholinesterase inhibitors. The results confirmed that increased diversity benefits MF-BO by enabling low-cost exploration, and there is an optimal level of correlation where the algorithm can effectively learn the utility of each fidelity.
- Figure 2D demonstrates that MF-BO is particularly effective at rediscovering the very top-ranked molecules, showing higher enrichment in the top 1% compared to the top 10% for the Factor-D data set.
- Experiments with decoy molecules added to the CXCR4 data set showed that MF-BO maintained its ability to rediscover active inhibitors, suggesting that biases from docking scores on inactive compounds do not significantly hinder the approach.
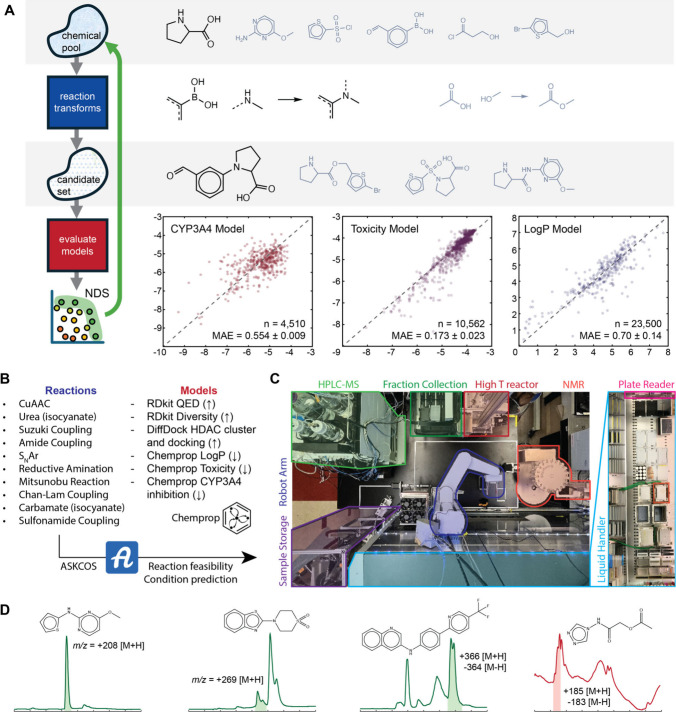
Figure 3. Schematic of molecular generation, reaction planning, reaction execution, and property testing in the MF-BO led discovery of new HDACIs. (A) The genetic algorithm for molecular generation selects building block molecules (top row) and applies reaction templates (middle row) to produce candidates (bottom row) iteratively. The candidates are ranked by several models, sorted by NDS, and top performers are recombined with building blocks to evolve more candidates. (B) Lists of the reactions and models used to generate candidates. ASKCOS is used to transform synthesis plans built during generation into executable experiments for the automated platform. (C) Top-down view of the automated platform. (D) Sample chromatograms of reactions yielding target products, as indicated by the expected mass-to-charge ratio, to demonstrate automated concentration analysis, with three passing reactions (green) and one failing (red) sample that could not be automatically deconvoluted.
Part 2: Prospective Application of MF-BO to Histone Deacetylase Inhibition
- A genetic algorithm (an optimization technique inspired by natural selection, used to find optimal solutions to problems by iteratively refining a population of potential solutions) was developed to generate a diverse search space of approximately 5,000 synthesizable and drug-like molecules, specifically excluding hydroxamic acids. Figure 3A provides a schematic of this molecular generation process.
- The autonomous platform integrated MF-BO to select molecules for docking (low fidelity), automated single-point percent inhibition assays (medium fidelity), and manual dose-response curve measurements (high fidelity).

Figure 4. (A) Timeline of automated discovery of HDACIs. Blue represents computational tasks, red represents manual tasks, and green represents automated physical experiments. Each green bar groups the actions required to prepare, process, and analyze a single plate of reactions. Cross hatched bars indicate pauses during which new operations were not started but operations underway were allowed to complete. The magenta box indicates an isolated equipment failure. The gray boxes represent weekends during which starting new work was suspended. Results of the first (B) and second (C) iterations of the prospective application of MF-BO to discover new HDACIs. A sample of generated molecules selected for docking, with docking rank and scores, are shown in pink. Some of the molecules automatically synthesized and tested for single-point percent inhibition are shown in blue, along with the distribution of percent inhibitions for the successfully synthesized molecules: gray bar indicates trichostatin A, a potent natural product, dark blue bars correspond to highlighted molecules with inset structures. Structures of molecules tested at the highest fidelity along with IC50 values are shown in green (cf. trichostatin A IC50 of 1.8 nM); note that in the first iteration, no dose–response experiments were proposed. (D) The retrosyntheses of the high-fidelity molecules in (C) along with reaction conditions and yield as determined by HPLC. Conditions. (a) dioxane, 100 °C, 4 h, 75% yield, 5:4 cis:trans isomers. (b) DCM, 20 °C, 1 h then 3 eq. sodium triacetoxyborohydride, 20 °C, 3 h, 42% yield. (c) 2 eq. acid chloride, dioxane, 50 °C, 1 h, then reflux, 16 h, 32% yield. (d) 3 eq. DIPEA, dioxane, reflux, 16 h, 5% yield. (e) 1 eq. hydrazine hydrate, THF, 20 °C, 15 min, quantitative yield. (f) 1 eq. Cu(OAc)2·H2O, 3 eq. pyridine, DCM, 20 °C, 4 h, air, 96% yield. (E) Dose–response curves from which the IC50 values in (C) were derived.
- Figure 4A shows the timeline of the two iterations of the discovery campaign, highlighting the computational tasks (blue), automated physical experiments (green), and manual tasks (red) involved. The pauses (hatched green) and the equipment failure (magenta) illustrate the real-world aspects of autonomous experimentation.
- In the first iteration, MF-BO prioritized docking and single-point assays. The platform attempted to synthesize 79 molecules, with 40 successful syntheses.
- Several tertiary sulfonamides showed appreciable inhibitory activity in the single-point screen. Figure 4B displays some of these selected candidates and their performance.
- The second iteration shifted the budget towards more dose-response assays. The platform attempted 106 single-point syntheses with 70 successes and manually synthesized 8 molecules for high-fidelity characterization. Figure 4C shows selected candidates from this iteration.
- Figure 4D presents the retrosyntheses of the successfully synthesized high-fidelity molecules (I-V) with their reaction conditions and yields. Figure 4E shows the dose-response curves for these molecules, revealing submicromolar inhibitors that do not contain hydroxamate moieties.
- While not as potent as trichostatin A (IC50 1.8 nM), some of the discovered molecules have IC50 values comparable to FDA-approved HDAC inhibitors like valproate and givinostat.
Conclusion
This research successfully developed and validated a multifidelity Bayesian optimization (MF-BO) algorithm that effectively accelerates the discovery of new drug molecules by strategically utilizing experimental data of varying fidelities. The study demonstrated the superiority of MF-BO over other common drug discovery approaches, particularly in diverse chemical search spaces with weak-to-moderate fidelity correlation.
The integration of MF-BO into an autonomous molecular discovery platform showcased the potential for fully automated and iterative drug discovery, leading to the identification of novel histone deacetylase inhibitors with desirable properties. The findings highlight the importance of considering both the cost and the information content of different experimental methods in drug discovery campaigns and provide a powerful framework for future automated molecular design and optimization efforts.


Automated Drug Discovery using Multifidelity Bayesian Optimization
Bayesian Optimization over Multiple Experimental Fidelities Accelerates Automated Discovery of Drug Molecules