The vast number of newly sequenced genomes presents a significant challenge: characterizing the functions of the encoded proteins.
Traditional methods for assigning protein function often rely on similarity to previously studied proteins but a huge number of unknown proteins remain a mystery. This approach creates an "annotation inequality," where well-studied proteins are easily annotated, while a vast universe of uncharacterized proteins remains unexplored.
The research presented here tackles this problem head-on by introducing EvoWeaver, a novel computational method designed to predict gene functional associations from coevolutionary signals.
By analyzing how genes have evolved together across diverse organisms, EvoWeaver aims to inject new information into our understanding of protein functions, particularly for those currently lacking annotations. This approach holds promise for accelerating the discovery of functions for the rapidly expanding collection of sequences.
Key Takeaways
- EvoWeaver provides a method to infer functional associations directly from sequencing data, without relying on existing knowledge or similarity to previously annotated proteins. This helps to combat the problem of annotation inequality and sheds light on those mysterious, unstudied proteins.
- Combines 12 different signals of coevolution from four primary categories: Phylogenetic Profiling, Phylogenetic Structure, Gene Organization, and Sequence Level methods. These signals look at which species have which genes, comparing gene evolutionary "family trees," checking gene location and organization, and analyzing changes in the gene sequences themselves. Combining these diverse signals leads to more accurate predictions.
- The method is designed for large-scale application, demonstrated by its use on 1545 gene groups from 8564 genomes, making it one of the largest coevolutionary analyses to date. Its predictions are more accurate than individual methods and incorporate statistical testing to reduce false associations.
- EvoWeaver is great at suggesting new ideas ("hypotheses") about how genes might work together, even identifying connections currently missing in major databases like KEGG and STRING.
- EvoWeaver is implemented as part of the SynExtend package for R, making it accessible to researchers.
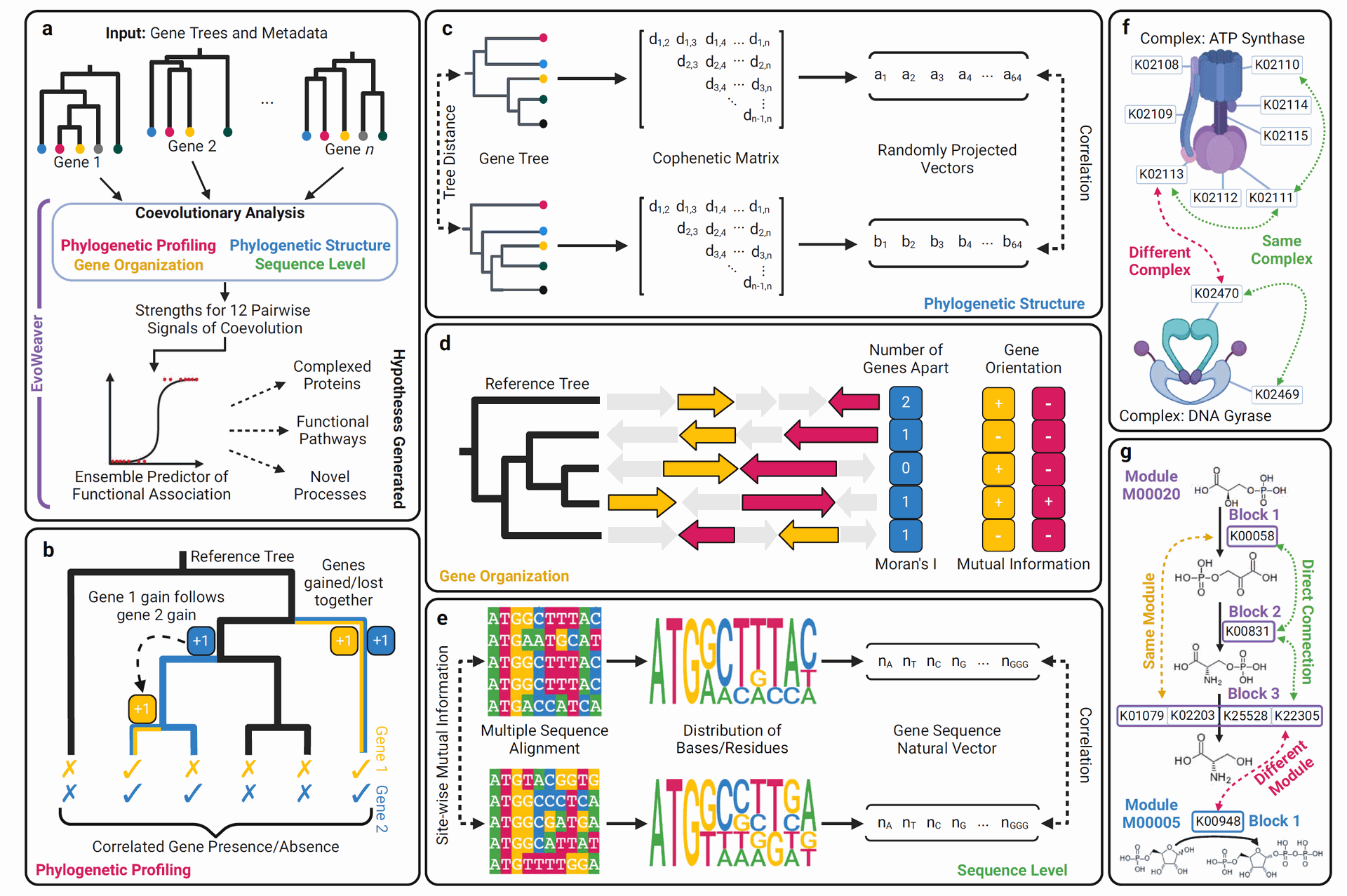
Fig. 1 | Overview of the EvoWeaver algorithm and benchmarking.
a Phylogenetic trees from groups of orthologous genes serve as the primary input to EvoWeaver. Four categories of coevolutionary signal are quantified for each pair of genes. These signals are combined in an ensemble classifier to predict functional relationships between gene pairs. EvoWeaver provides as output its 12 predictions for signals of coevolution, and can optionally provide an ensemble prediction using built-in pretrained models. b Functional associations often result in correlated gain/loss patterns on a reference phylogenetic tree (e.g., a species tree). EvoWeaver assesses the presence/absence patterns, correlation between gain/loss events, and distance between gain/loss events as signals of coevolution. c Similarity in phylogenetic structure is another indicator of coevolution between genes. EvoWeaver computes topological distance as well as correlation in patristic distances following dimensionality reduction using random projection. d Functionally associated genes sometimes cluster on the genome due to coregulation or horizontal gene transfer. EvoWeaver derives signals from the conservation in gene orientation and the distance between gene pairs. e Functional associations sometimes cause concerted changes in sequences that are interrogated by EvoWeaver. EvoWeaver can analyze nucleotide sequences or amino acid sequences, though nucleotide sequences are pictured here. f Proteins involved in the same complex are functionally associated and can be identified through signals of coevolution. The goal of the Complexes benchmark is to distinguish orthology groups in the same complex (i.e., positives) from those in different complexes (i.e., negatives). g Functional associations between proteins that are adjacent in the same module are stronger than those between different modules. The goal of the Modules benchmark is to distinguish adjacent proteins in the same module from independent modules. Created in BioRender. Lakshman, A. (2025)
Overview
The rapid growth of genome sequencing has created a significant gap between the number of known protein sequences and the number of proteins with experimentally determined functions.
Computational annotation methods, while helpful, often rely on sequence similarity to known proteins, meaning that proteins without close homologs to characterized proteins remain unannotated.
This contributes to annotation inequality, where certain areas of the "protein universe" are well-lit, while others remain in the dark.
A promising approach to infer protein function is through coevolution. The principle of "guilt-by-association" suggests that genes whose products work together in a cell (e.g., in the same protein complex or biochemical pathway) are often under shared selective pressure and tend to evolve together. This shared evolutionary history leaves detectable molecular signals in their genetic code and organization.
Historically, coevolutionary analyses have used four primary approaches:
- Phylogenetic Profiling: Examining the presence or absence of genes across different genomes. Genes that are functionally linked are often gained or lost together.
- Phylogenetic Structure: Comparing the evolutionary histories (phylogenetic trees) of different genes. Functionally linked genes tend to have similar evolutionary trees.
- Gene Organization: Analyzing the physical location and orientation of genes on the genome. Genes involved in the same pathway or complex are sometimes clustered or oriented similarly.
- Sequence Level Methods: Looking for correlated changes in the amino acid or nucleotide sequences of genes. Specific residues in interacting proteins might co-vary to maintain functional compatibility. For example, if two proteins physically interact, changes in one might require a corresponding change in the other to keep them working together.
However, existing coevolutionary algorithms often face challenges with accuracy and scalability, making it difficult to apply them to the entire universe of proteins. These four approaches have largely been applied independently limiting the predictive power of the associations across the different types of coevolutionary signals.
EvoWeaver addresses these limitations by weaving together 12 different algorithms that capture these diverse coevolutionary signals.
The tool takes phylogenetic gene trees and optional metadata (like genomic location or sequences) as input. It then calculates a score between -1 and 1 for each pair of gene groups using each of the 12 algorithms, quantifying the strength of their coevolution.
These scores are then combined using ensemble machine learning models (logistic regression, random forest, or neural network) to generate a final prediction about the functional association between the gene pairs. This system then provides a final "best guess" about whether the gene pairs are functionally linked for the user to evaluate.
Why it’s Important
EcoWeaver is helping to overcome annotation bias by predicting functional associations de novo from sequence data without relying on existing annotations or text mining of literature, EvoWeaver can shed light on poorly studied proteins and help reduce annotation inequality. This is crucial because biases in protein function annotation exist and can hinder research. Many potentially important genes are currently overlooked.
To overcome computational limitations of single algorithms, EvoWeaver is designed to be highly scalable, with optimized algorithms and pairwise comparisons that are independent and can be distributed across computing clusters. This allows it to analyze very large datasets, far exceeding the scale of many previous coevolutionary analyses.
The case study of human genes B3GNT5 and ST6GAL1 (Fig. 5) exemplifies this, where EvoWeaver predicted a connection not present in KEGG or STRING, which was later supported by experimental evidence showing B3GNT5 modulates ST6GAL1 expression. EvoWeaver is positioned as a powerful tool for generating hypotheses about functional associations that can then be tested experimentally providing researchers with new targets and understanding of relationships.
Finally, EvoWeaver can not only predict associations but also classify their strength into hierarchical levels (Direct Connection, Same Module, Same Pathway, etc.), providing more nuanced insights into functional relationships.
EvoWeaver doesn't just say "yes, they're connected." It can also estimate how connected they are, sorting links into levels like "Direct Connection" (they work right next to each other), "Same Module" (they're part of the same small team or process step), or "Same Pathway" (they're part of the same larger process). This gives us a more detailed picture of functional relationships across genes.
Beyond protein-coding genes, the authors anticipate that EvoWeaver's predictions could potentially be useful for other sequence features, such as non-coding RNAs, although this was not the focus of the study.
Summary of Results
The researchers developed EvoWeaver as a scalable software package within the R environment, available via Bioconductor as part of the SynExtend package.
EvoWeaver's core function is to take phylogenetic gene trees and optional metadata, perform 12 different coevolutionary analyses across four categories, and output scores for each gene pair.
These scores are then fed into machine learning classifiers (logistic regression, random forest, neural network) trained on data with known functional associations to make ensemble predictions.
To evaluate EvoWeaver, several benchmark datasets based on the KEGG database were constructed. KEGG provides a hierarchical structure of protein complexes, modules (building blocks of pathways), and pathways.
Benchmarking and Performance:
- Complexes Benchmark: Tested EvoWeaver's ability to distinguish pairs of genes encoding proteins in the same complex (positives) from unrelated pairs (negatives). This benchmark assessed the prediction of direct physical interactions.
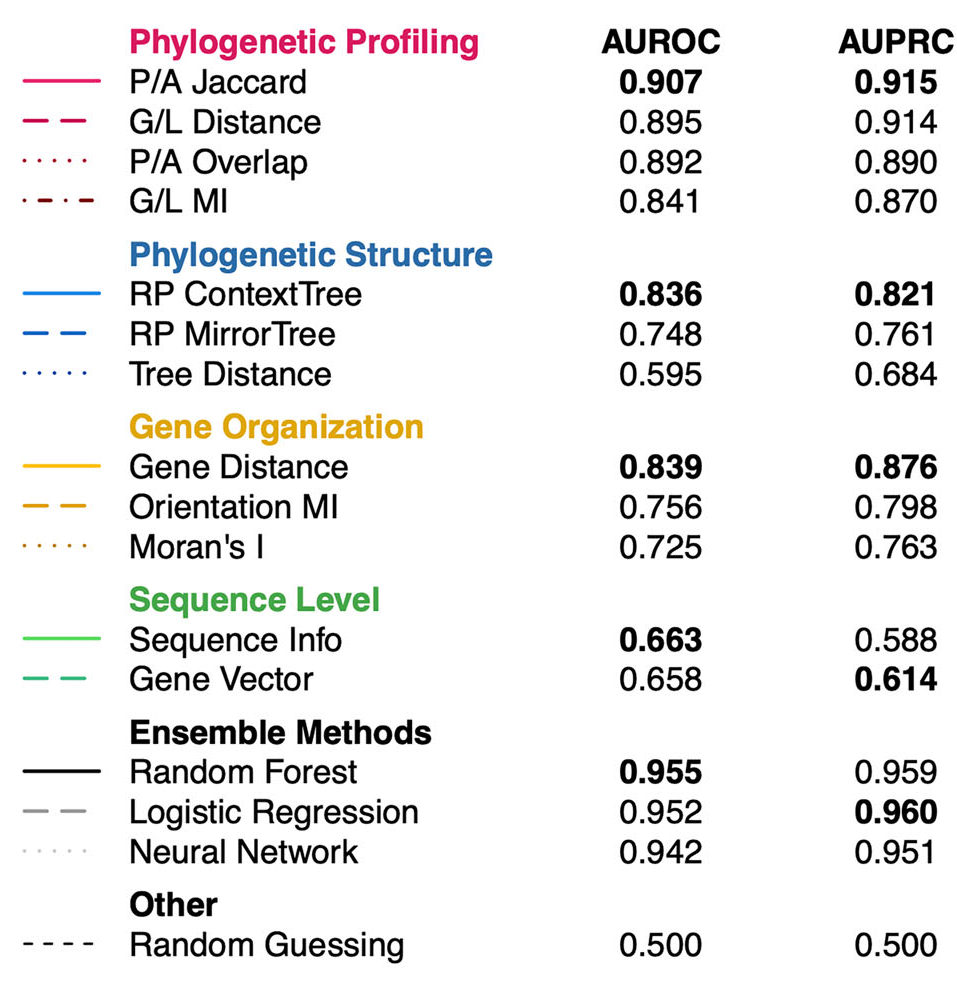
Figure 2. EvoWeaver’s ensemble predictions outperform individual algorithms on the Modules benchmark.
- Modules Benchmark: Evaluated the ability to distinguish adjacent proteins within KEGG modules (positives) from proteins in distinct modules (negatives). Figure 2 shows the performance on this benchmark, demonstrating that while individual algorithms vary in accuracy, EvoWeaver's ensemble predictions significantly outperform them.
- Multiclass Benchmark: Explored EvoWeaver's ability to predict hierarchical levels of functional association defined by KEGG: Direct Connection, Same Module, Same Pathway, Same Global Pathway, and Unrelated. Using a Random Forest model, EvoWeaver demonstrated high accuracy in classifying these relationships.
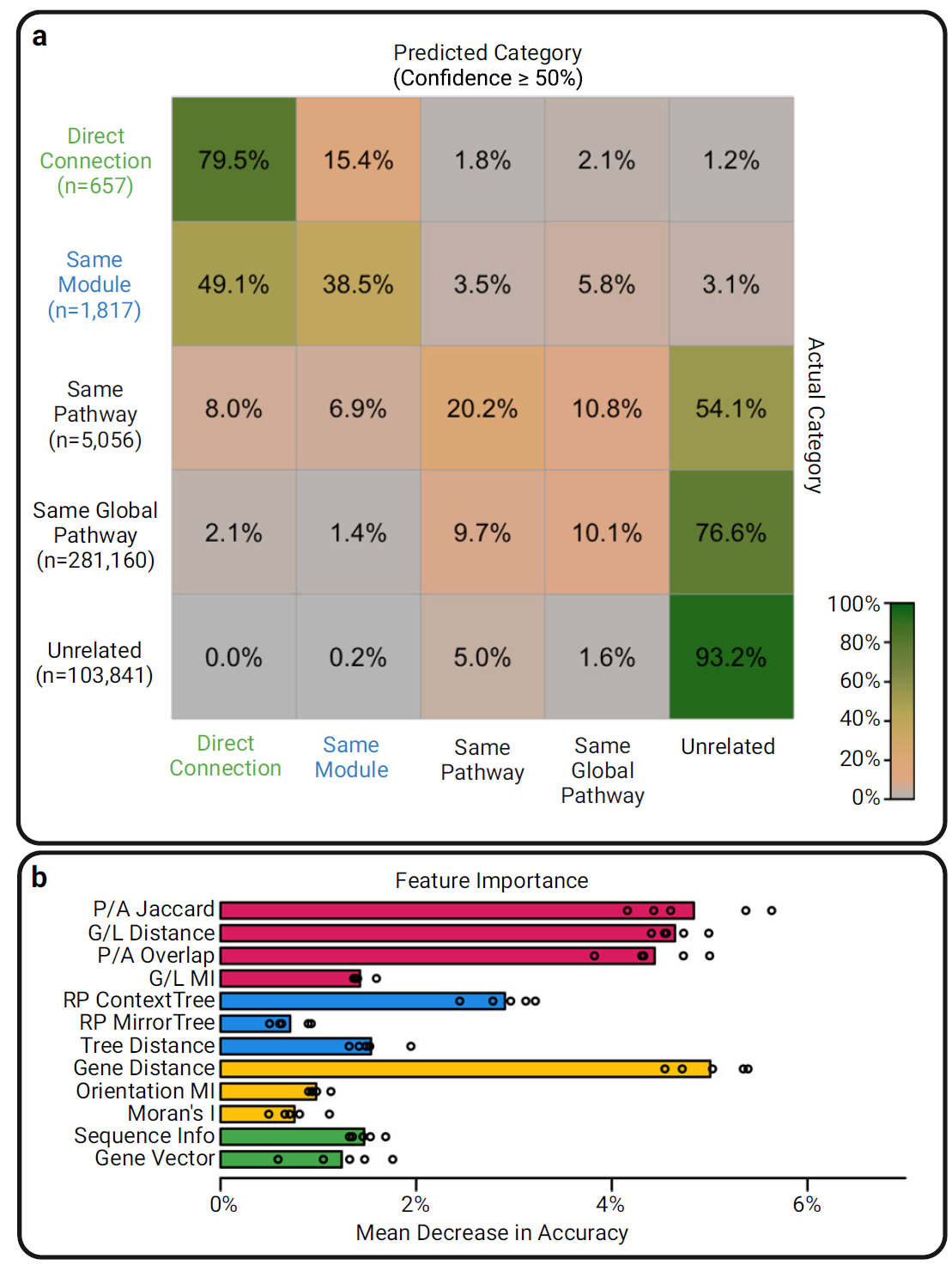
Figure 3a presents the confusion matrix, showing that predictions rarely confused genes within the same module with those from different modules. Figure 3b indicates that all 12 component predictors contributed to the ensemble classifier's accuracy in this task, reinforcing the benefit of combining multiple signals.
Case Studies and Hypothesis Generation:
The case study of human genes B3GNT5 and ST6GAL1 demonstrated EvoWeaver's ability to predict a functional association (Direct Connection/Same Module) not present in KEGG or STRING.
This prediction was supported by phylogenetic profiling, gene organization, phylogenetic structure, and sequence-level evidence (Fig. 5c-f) and is consistent with experimental findings showing a regulatory link between the two genes.
Looking closely at cases where EvoWeaver was very confident in its prediction, but the prediction turned out to be "wrong" according to the KEGG database, actually revealed some interesting biological insights.
For example, many pairs of genes that KEGG listed as just being in the "Same Pathway" were predicted by EvoWeaver to have a "Direct Connection." When scientists checked, many of these pairs were indeed directly linked or were very close steps in the process. This suggests that some of EvoWeaver's "wrong" predictions might actually be finding connections that are missing or not fully detailed in KEGG!
In another case, two groups of genes involved in related biological processes (making certain sugar molecules) were listed by KEGG as only being in the "Same Global Pathway." But EvoWeaver predicted they had a "Direct Connection."
Both coevolutionary signals and lab experiments support that these gene groups are indeed closely linked. This further shows how EvoWeaver can suggest connections that databases haven't explicitly written down yet.
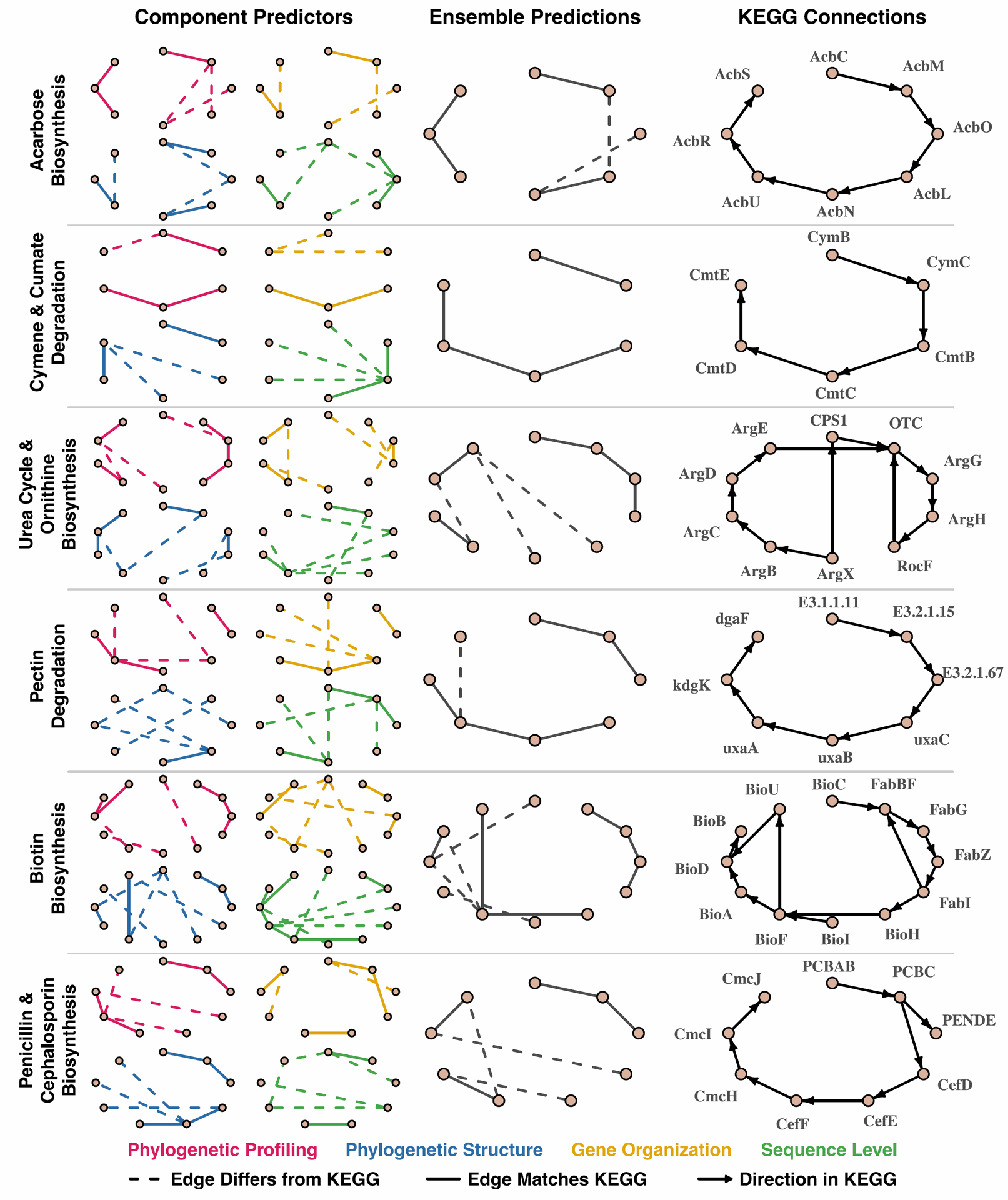
Fig. 6 | EvoWeaver partly identifies biochemical pathway connectivity.
Figure 6 gives a visual example, showing how EvoWeaver partly figures out connections within biological processes by highlighting the strongest predicted links between genes in example pathways from KEGG.
While it correctly finds many real connections (shown as solid lines), it sometimes incorrectly links genes that are next to each other in a process (dashed lines). This shows that EvoWeaver is great at finding that genes are linked, but it doesn't necessarily tell you the order or direction of the steps in the process.
Overall, the results demonstrate that EvoWeaver is a powerful, accurate, and scalable tool for predicting gene functional connections, especially for proteins we know little about, and for generating exciting new ideas for biological research.
Conclusion
EvoWeaver represents a significant advancement in the field of computational gene function prediction, leveraging the power of coevolutionary signals to uncover functional associations on a large scale.
By moving beyond methods reliant on similarity to known proteins or existing database annotations, EvoWeaver directly addresses the growing challenge of annotation inequality for the vast universe of uncharacterized proteins.
The key innovation lies in the integration of 12 distinct coevolutionary algorithms across four levels of biological organization (organism, genome, gene, sequence) and the use of ensemble machine learning to combine these signals.
This multi-faceted approach allows EvoWeaver to capture a more comprehensive view of coevolution, leading to improved accuracy compared to methods relying on single signals. The design prioritizes scalability, enabling analysis of thousands of genomes and millions of genes, a critical feature for modern biological data.
The researchers see EvoWeaver not as something to replace lab experiments or existing databases, but as a great tool for generating new ideas ("hypotheses") for scientists to test. The fact that it can predict connections not found in big databases like KEGG and STRING, and that some of its seemingly "wrong" predictions are actually supported by lab evidence, highlights its potential to guide future research and uncover biological relationships we didn't know about before.
Implementing EvoWeaver within the R SynExtend package makes this powerful tool accessible to the scientific community. As sequencing data continues to accumulate rapidly, tools like EvoWeaver will be indispensable for unlocking the functional secrets of the protein universe and driving new discoveries in biology and medicine.

Machine Learning Helps in Predicting What Genes Do and How They Work Together
EvoWeaver: large-scale prediction of gene functional associations from coevolutionary signals